СЕМИОТИКА ВРОЖДЕННЫХ И НАСЛЕДСТВЕННЫХ СИНДРОМОВ Генетика человека изучает явления наследственности и изменчивости в популяциях человека, особенности наследования признаков в норме и изменения их под действием условий окружающей среды. Задачей медицинской (клинической) генетики является разработка методов диагностики, лечения и профилактики наследственных болезней человека. Прогресс в развитии медицины приводит к относительному возрастанию доли генетически обусловленной патологии. К настоящему времени описано свыше 3500 наследственных болезней. Около 5—5,5 % детей рождаются с наследственной или врожденной патологией. Половина спонтанных абортов обусловлена генетическими причинами. Не менее 30 % перинатальной и нео-натальной смертности обусловлено врожденными пороками развития и наследственными болезнями с другими проявлениями. С генетической точки зрения все болезни в зависимости от роли наследственных и средовых факторов в их развитии можно подразделить на 3 группы. 1. Наследственные болезни — фенотипическое проявление мутации как этиологического фактора практически не зависит от среды; последняя может только изменять выраженность симптомов и тяжесть течения болезни. Это генные и хромосомные наследственные болезни (гемофилия, фенилкетонурия, муко-висцидоз, болезнь Дауна и др.). 2. Болезни с наследственной предрасположенностью. Их в свою очередь можно подразделить еще на два вида. Болезни, наследственность при которых является этиологическим фактором, но для их проявления необходимо действие соответствующего фактора внешней среды (например, подагра, диабет). Болезни, этиологическими факторами при которых являются средовые влияния, однако частота возникновения и тяжесть течения болезней зависят от наследственной предрасположенности. К таким болезням относятся атеросклероз, гипертоническая болезнь, язвенная болезнь, псориаз и др. 3. Болезни, в происхождении которых наследственность не играет роли. Это, например, травмы, ожоги, инфекционные болезни. Генетические факторы в этом случае могут влиять только на течение патологических процессов (скорость регенерации, выздоровления, компенсации функций). Мутации как этиологический фактор. Этиологическими факторами наследственных болезней являются геномные, хромосомные и генные мутации. Заболевания, обусловленные изменениями числа и структуры хромосом (геномные и хромосомные мутации соответственно), называются хромосомными болезнями. При хромосомных болезнях нарушается сбалансированность набора генов и наблюдаются отклонения от нормального развития организма. Это приводит к внутриутробной гибели эмбрионов и плодов, врожденным порокам развития и другим клиническим проявлениям. При хромосомных болезнях отклонения от нормального развития коррелируют, как правило, со степенью хромосомного дисбаланса. Чем больше хромосомного материала вовлечено в мутацию, тем раньше проявляется заболевание и тем значительнее нарушения в физическом и психическом развитии индивидуума. Избыток генетического материала проявляется, как правило, легче, чем его недостаток. Заболевания, обусловленные изменениями структуры молекулы ДНК (генные мутации), называются генными болезнями. Фенотипически генные мутации могут проявляться на молекулярном, клеточном, тканевом, органном и организменном уровнях. Генные мутации наследуются по законам Менделя. Для наследственных болезней характерен клинический полиморфизм. Он обусловлен взаимодействием генетических и средо-вых факторов. Важное место в этом феномене занимают такие генетические явления, как генетическая гетерогенность организмов, пенетрантность и экспрессивность генов, явления плей-отропии и взаимодействие аллельных и неаллельных генов. Термины «наследственные болезни» и «врожденные болезни» не являются синонимами, так как врожденные болезни (проявляющиеся с момента рождения) могут быть обусловлены как наследственными, так и средовыми тератогенными факторами (талидомид, сифилис, краснуха). В то же время не все наследственные болезни являются врожденными (вероятно, их около 50 %). Некоторые болезни проявляются в детском (миопатия Дюшенна, гемофилия), другие — в зрелом (миотоническая ди-стония, хорея Гентингтона) и даже в пожилом (болезнь Альцгеймера) возрасте. В основу генетической классификации наследственных болезней положен этиологический принцип: тип мутаций и характер взаимодействия со средой. Наследственные болезни подразделяются на 5 групп: генные болезни, хромосомные болезни, болезни с наследственной предрасположенностью (мультифакториальные), генетические болезни соматических клеток и болезни генетической несовместимости матери и плода. Болезни с наследственной предрасположенностью могут быть моногенными и полигенными. Генетические болезни соматических клеток выделены в самостоятельную группу наследственной патологии недавно. Они связаны с изменениями генетического материала соматических клеток, активирующими онкогены, вызывающими аутоиммунные процессы и др. Болезни при несовместимости матери и плода по антигенам возникают в результате иммунологической реакции материнского организма на антигены плода. Наиболее хорошо изученным заболеванием этой группы является гемолитическая болезнь новорожденных, развивающаяся вследствие несовместимости матери и плода по Rh-антигенам. Эта группа составляет около 1% патологии новорожденных. Особенности клинических проявлений наследственной патологии: 1. Наследственные заболевания часто носят семейный характер. В то же время наличие заболевания только у одного из членов родословной не исключает наследственного характера этой болезни (новая мутация, появление рецессивной гомозиготы). Общие принципы клинической диагностики наследственных болезней. Диагностика наследственной патологии является сложным и трудоемким процессом. Трудности обусловлены большим количеством нозологических форм (3500), разнообразием клинической картины каждой из них, наличием фенокопий, редкой встречаемостью некоторых форм. Врач общей практики не может владеть всем запасом знаний, необходимых для диагностики наследственных болезней, поэтому знание основных принципов диагностики этой патологии является целесообразным. Клиническая диагностика наследственных болезней основывается На данных клинического, генеалогического и параклинического обследования. Следует помнить, что наследственные болезни могут протекать сходно с ненаследственными. В некоторых случаях наследственная патология может сопутствовать основному, ненаследственному, заболеванию. Поэтому постановка диагноза должна быть двухэтапной: 1) общее клиническое обследование больного в соответствии с современными требованиями; Общие клинические методы часто являются основными в диагностике наиболее известных и распространенных наследственных болезней (например, болезни Дауна). Полного клинического и параклинического обследования обычно достаточно для диагностики таких наследственных заболеваний, как ахонд-роплазия, нейрофиброматоз, хорея Гентингтона и др. Однако при этом возможны диагностические ошибки. Для их исключения, как правило, в дальнейшем необходимо проведение специальных генетических методов обследования (изучение кариоти-па, структуры молекулы ДНК и др.). Особенности обследования больного с наследственной патологией. Вначале рассмотрим наиболее очевидные признаки наследственной патологии — врожденные пороки развития. Под врожденными пороками развития понимаются стойкие морфологические изменения тканей или органов, выходящие за пределы вариаций их строения. Формирование врожденных пороков развития в результате нарушения нормального течения эмбриогенеза называется тератогенезом (teras, teratos — урод, чудовище). Наука об этиологии, патогенезе и проявлениях врожденных пороков развития называется тератологией. Частота грубых врожденных пороков развития, сопровождающихся нарушением функций, в популяциях человека составляет 2—3 %. Формирование врожденных пороков — результат отклонений от нормального развития особи. Единый процесс онтогенеза слагается из следующих этапов: 1) гаметогенез; В результате гаметогенеза образуются половые клетки, несущие в себе генетическую информацию, в процессе реализации которой из одной клетки (зиготы) развивается многоклеточный организм. При оплодотворении происходит объединение генетической информации материнского и отцовского организмов, что обусловливает комбинативную изменчивость. Формирование морфологических структур эмбриона (эмбриональный морфогенез) включает эмбриональный гистогенез (возникновение специализированных тканей из малодифференцированных клеток эмбриональных зачатков) и органогенез (развитие органов и систем органов). Эмбриональный морфогенез осуществляется при взаимодействии генотипа зародыша и организма матери и связан с процессами размножения, роста, дифференцировки, миграции и отмирания клеток. Эти процессы контролируются сложными взаимодействиями генетических, эпигеномных и внешних факторов, определяющих в конечном итоге временную и пространственную последовательность экспрессии (включения и выключения) блоков генов и тем самым дифференцировку клеток и морфогенез. Нарушение в процессе эмбриогенеза любого из вышеперечисленных механизмов вызывает отклонение от нормального развития, что может реализоваться во врожденном пороке. На внутриклеточном уровне к «пусковым» механизмам нарушения развития относятся изменения молекулярных процессов репликации ДНК, биосинтеза белков-ферментов и др. К основным клеточным механизмам тератогенеза относятся нарушения размножения, миграции и дифференцировки клеток. Результатом снижения митотической активности клеток могут быть гипоплазии или аплазии органа или его части. В результате нарушения миграции клеток могут развиваться гетеро-топии и другие пороки. Дифференцировка, то есть образование разнородных клеток, тканей и органов из однородного эмбрионального зачатка, происходит последовательно в течение всего эмбриогенеза. Основной механизм специализации клеток — дифференциальная активность генов, в результате которой в разные фазы эмбриогенеза синтезируются специфические для каждой стадии ферменты, которые в основном и обеспечивают специализацию клеток. Если нарушается механизм включения и выключения отдельных блоков генов, это приводит к развитию различных пороков, К основным механизмам тератогенеза на тканевом уровне относятся гибель отдельных клеточных масс, замедление распада и рассасывания клеток, отмирающих в ходе нормального эмбриогенеза, и нарушение адгезии тканей. Физиологическая гибель клеток происходит под действием лизосомальных ферментов в процессе окончательного формирования органов. Такая первичная гибель клеток наблюдается при слиянии нёбных отростков, открытии естественных отверстий, формировании пальцев. Задержка или замедление физиологического распада клеток могут приводить к синдактилии, атрезиям и другим порокам развития. Так как пороки развития чрезвычайно многообразны, классификация их затруднена. Наиболее распространены классификации по этиологическому принципу и по локализации. По этиологическому признаку различаются три группы пороков: наследственные, экзогенные и мультифакториальные. К наследственным относятся пороки, возникшие в результате мутаций. В зависимости от вида изменений генетического материала наследственно обусловленные пороки подразделяются на генные и хромосомные. В группу экзогенных объединяются пороки, обусловленные действием тератогенных факторов непосредственно на эмбрион или плод. Пороки мультифакториальной (комбинированной) этиологии являются результатом совместного воздействия генетических и экзогенных факторов. В настоящее время считается (Н.П. Бочков, 1997), что из общего числа врожденных пороков развития генетически обусловленные формы составляют 20—30 %, мультифакториальные — 30—40 %, экзогенные — 2—5 %, неясной этиологии — 25—50 %. Условность приведенной этиологической классификации очевидна. Вместе с тем для медико-генетического прогноза важно знать, какой из факторов является ведущим (генетический или средовый). По объекту воздействия тератогенных факторов врожденные пороки могут быть разделены на гаметопатии, бластопатии, эмбриопатии и фетопатии. Гаметопатии — это наследственно обусловленные врожденные пороки, в основе которых лежат мутации в половых клетках родителей (например, синдром Дауна, обусловленный свободной трисомией; все врожденные пороки, обусловленные новой доминантной мутацией). Бластопатиями называются поражения бластоцисты, то есть зародыша первых 15 дн после оплодотворения. Следствием бластопатий являются двойниковые пороки, циклопия и др. Эмбриопатии — это врожденные пороки, возникшие в результате повреждения эмбриона (с 16-го дня после оплодотворения до конца 8-й недели). К последствиям эмбриопатий относятся диабетические, алкогольные, медикаментозные и многие другие пороки. Фетопатии — это повреждения плода (от 9-й недели дб рождения). Пороки этой группы сравнительно редки. К ним можно отнести крипторхизм, гипоплазии и др. По распространенности в организме врожденные пороки подразделяются на изолированные (одиночные), локализованные в одном органе (стеноз привратника), системные — в пределах одной системы органов (хондродисплазия) и множественные — в органах двух и более систем (сочетание расщелины губы, косолапости и порока сердца). Наиболее распространена классификация изолированных врожденных пороков развития, основанная на анатомо-физиологических принципах деления тела человека на системы органов (пороки ЦНС и органов чувств, сердечно-сосудистой системы и т.д.). Множественные пороки чаще классифицируются по этиологическому принципу (хромосомные синдромы, генные синдромы, синдромы, обусловленные экзогенными факторами, и др.). Важным этапом при обследовании больного с клинико-ге-нетической точки зрения является антропометрия. Для диагностики наследственных болезней важными оказываются следующие антропометрические данные: рост, масса тела, телосложение, пропорции тела, длина конечностей, длина туловища, окружность груди и черепа и др. При осмотре пациентов наряду с выявлением врожденных пороков развития и проведением антропометрического обследования необходимо обращать внимание на микроаномалии развития, или врожденные морфогенетические варианты (отклонения в развитии, которые выходят за пределы нормальных вариаций, но не нарушают функции органа). Они являются неспецифическими показателями эмбрионального дисморфогенеза. Врожденные морфогенетические варианты встречаются у людей без какой-либо врожденной или наследственной патологии, но наличие более 5 таких признаков у одного индивидуума указывает на необходимость тщательного обследования его на предмет врожденной или наследственной патологии. Наиболее распространенные признаки дисморфогенеза, учитываемые при дифференциальной диагностике наследственных болезней: 1) кожа: ангиомы, телеангиэктазии, пигментные пятна, веснушки темные (свыше 20), депигментация, гипертрихоз, гирсутизм, липомы, фибромы, келоидные рубцы, нарушение потоотделения, ихтиоз, повышенная растяжимость; Существенным признаком, указывающим на наследственную или врожденную патологию, является нарушение течения беременности и пренатального развития плода (прерывание беременности, мало- и многоводие, несоответствие размеров и массы плода и новорожденного и др.). Синдромологический подход к диагностике наследственных болезней. Известно, что в наследственной патологии не существует патогномоничных признаков. Чаще всего один и тот же признак встречается при многих заболеваниях, например аномалии почек — при 30, а искривление позвоночника — при 50 наследственных синдромах. При внимательном осмотре больного врач может выявить признаки, существенно облегчающие дифференциальную диагностику. Например, запавшая переносица может быть признаком мукополисахаридоза и ахондроплазии; искривление нижних конечностей — следствием не только рахита, но и 25 различных наследственных болезней. Умственная отсталость — результат патологии при более чем 100 наследственных синдромах. Наследственные формы часто встречаются в практике окулистов. Атрофия зрительных нервов наблюдается при 15, катаракта и помутнение хрусталика — более чем при 30 наследственных болезнях. Параклинические исследования играют существенную роль в диагностике наследственных болезней. Например, клинико-биохимические исследования проводятся при муковисцидозе, семейной гиперхолестеринемии, болезни Вильсона-Коновалова. Гематологические исследования позволяют подтвердить гемоглобинопатии, гемофилии. Эндокринологические исследования проводятся при врожденном гипотиреозе, адреногенитальном синдроме, нанизме. Электрофизиологические исследования позволяют установить различные виды миодистрофий и наследственную патологию нервной системы. Методы изучения генетики человека. Изучение генетики человека связано с рядом особенностей и объективных трудностей: 1) сложный кариотип; Для обследования больных и решения вопросов патогенеза наследственных заболеваний в медицинской генетике широко применяются общеклинические методы: электрокардиография, электроэнцефалография, электромиография, биохимические анализы биологических жидкостей, биопсия тканей и др. Однако имеется целый ряд специфических методов, с помощью которых можно изучить вопросы возникновения, развития, распространения, механизмы передачи из поколения в поколение наследственных болезней и роль генотипа и факторов среды в их проявлении. Клиникогенеалогический метод был предложен в 1883 г. Ф. Гальтоном. Он основан на построении родословных и прослеживании в ряду поколений передачи определенного признака. Этот метод относится к наиболее универсальным методам медицинской генетики. Он широко применяется для решения теоретических и прикладных проблем. Метод позволяет установить: 1) является ли данный признак наследственным (по проявлению его у родственников); Этапы генеалогического анализа: 1) сбор данных обо всех родственниках обследуемого (анамнез); Обычно родословная собирается по одному или нескольким признакам. В зависимости от цели исследования родословная может быть полной или ограниченной. Желательно стремиться к наиболее полному составлению родословной по восходящему, нисходящему и боковым направлениям. Сложность сбора анамнеза заключается в том, что пробанд должен хорошо знать родственников по линии матери и отца не менее трех поколений и состояние их здоровья, что бывает крайне редко. Одного опроса, как правило, недостаточно. Для некоторых членов родословной приходится назначать полное клиническое, параклиническое или лабораторное обследование для уточнения состояния их здоровья. Подробное клинико-генеалогическое исследование проводится во всех случаях, когда при первичном клиническом осмотре возникает подозрение на наследственную болезнь. Для построения родословных применяются условные обозначения. При построении родословной необходимо соблюдать следующие правила: 1) родословную начинают строить с пробанда;
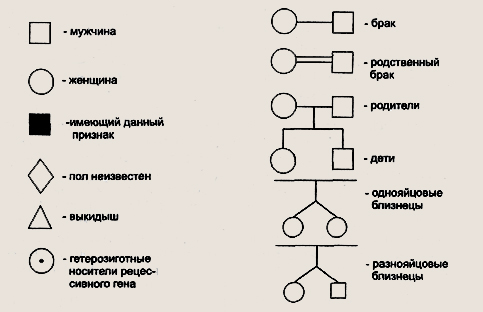
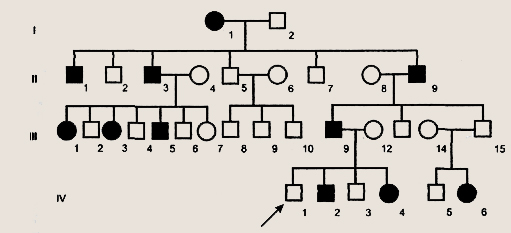
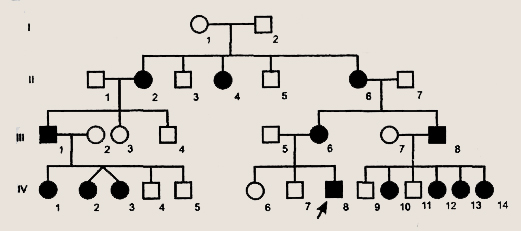
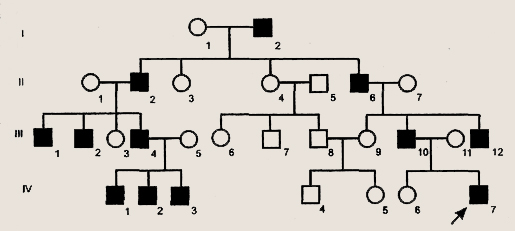
Условные обозначения для построения родословных Первая задача при анализе родословной — установление наследственного характера признака. Если в родословной встречается один и тот же признак (болезнь) несколько раз, то можно думать о его наследственной природе. После обнаружения наследственного характера признака (болезни) необходимо установить тип наследования. Для этого используются принципы генетического анализа и различные статистические методы обработки данных многих родословных. Аутосомно-доминантный тип наследования характеризуется следующими признаками: 1) больные в каждом поколении; Доминантно наследуемые болезни характеризуются полиморфизмом клинических проявлений и высокой вариабельностью сроков начала болезни. Для большинства болезней этого типа характерны такие патологические состояния, которые не наносят серьезного ущерба здоровью человека и в большинстве случаев не влияют на его способность иметь потомство. Так наследуются у человека полидактилия (шестипалость), веснушки, курчавые волосы, нейрофиброматоз , ахондроплазия, синдром Марфана и др. Следует подчеркнуть, что перечисленные признаки аутосомно-доминантного типа наследования будут проявляться только при полном доминировании. При неполном доминировании у потомков будет проявляться промежуточная форма наследования. При неполной пенетрантности гена больные могут быть не в каждом поколении.
Родословная с аутосомно-доминантным типом наследования
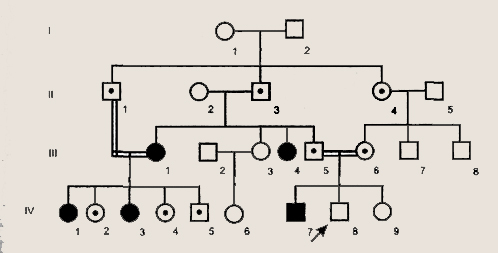
Родословная с аутосомно-рецессивным типом наследования Аутосомно-рецессивный тип наследования характеризуется следующими признаками: 1) больные не в каждом поколении;
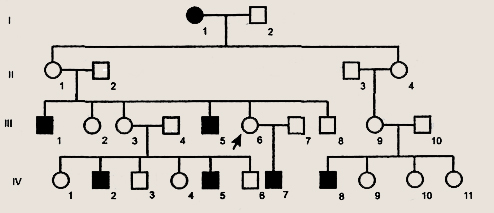
Родословная с Х-сцепленным рецессивным типом наследования Х-сцепленный рецессивный тип наследования характеризуется следующими признаками: 1) больные появляются не в каждом поколении; Х-сцепленный доминантный тип наследования сходен с аутосомно-доминантным, за исключением того, что мужчина передает этот признак только дочерям (сыновья получают от отца Y-хромосому). Примером такого заболевания является особая форма рахита, устойчивая к лечению витамином D. Голандрический тип наследования характеризуется следующими признаками: 1) больные во всех поколениях; Так наследуются у человека некоторые формы ихтиоза, обволошенность наружных слуховых проходов и средних фаланг пальцев, некоторые формы синдактилии (перепонки между пальцами ног) и др.
Родословная с Х-сцепленным доминантным типом наследования
Родословная с голандрическим типом наследования Близнецовый метод изучения генетики человека введен в медицинскую практику Ф. Гальтоном в 1876 г. Он позволяет определить роль генотипа и среды в проявлении признаков. Различают моно- и дизиготных близнецов. Монозиготные (однояйцовые) близнецы развиваются из одной оплодотворенной яйцеклетки. Монозиготные близнецы имеют совершенно одинаковый генотип, но могут отличаться по фенотипу, что обусловлено воздействием факторов внешней среды. Дизиготные (двуяй-цовые) близнецы развиваются после оплодотворения сперматозоидами нескольких одновременно созревших яйцеклеток. Такие близнецы имеют разный генотип, и их фенотипические отличия обусловлены как генотипом, так и факторами внешней среды. Монозиготные близнецы имеют большую степень сходства по признакам, которые определяются в основном генотипом. Например, они всегда однополы, у них одинаковые группы крови по разным системам (ABO, Rh, MN и др.)> одинаковый цвет глаз, однотипные дерматоглифические узоры на пальцах и ладонях и др. Эти фенотипические признаки и используются в качестве критериев диагностики зиготности близнецов. Процент сходства близнецов по изучаемому признаку называется конкордантностью, а процент различия — дискордант-ностью. Так как монозиготные близнецы имеют одинаковый генотип, то конкордантность у них выше, чем у дизиготных. Для оценки роли наследственности и среды в развитии того или иного признака используется формула Хольцингера: КМБ(%) - КДБ(%) /100%-КДБ(%) где Н — доля наследственности, КМБ — конкордантность монозиготных близнецов, КДБ — конкордантность дизиготных близнецов. Если результат расчетов по формуле Хольцингера приближается к единице, то основная роль в развитии признака принадлежит наследственности, и наоборот, чем ближе результат к нулю, тем больше роль средовых факторов. Популяционно-статистический метод изучения генетики человека основан на использовании математического выражения закона Харди—Вейнберга, где р — частота встречаемости в популяции доминантного гена, q — частота встречаемости рецессивного гена, р2 — частота доминантных гомозигот, q2 — частота рецессивных гомозигот, a 2pq — частота гетерозигот. Сумма частот всех генотипов может быть принята за 1 (100 %): р2 + 2pq + q2 = 1(100 %). Метод позволяет определять частоту генов и генотипов в больших (свыше 4,5 тыс.) популяциях людей. Цитогенетические методы. Цитогенетический метод основан на макроскопическом исследовании кариотипа. Этапы исследования: 1) культивирование клеток человека (чаще лимфоцитов) на искусственных питательных средах; В 70-е годы прошлого века были разработаны методы дифференциального окрашивания хромосом человека, которые показали, что каждая пара хромосом имеет свой специфический характер чередования неокрашенных, светло- и темноокрашенных дисков (Парижская классификация). Метод позволяет выявлять геномные (например, болезнь Дауна) и хромосомные (например, синдром кошачьего крика) мутации. В таких случаях кариотип больного обозначают следующим образом: количество хромосом, набор гетерохромосом, номер хромосомы, короткого или длинного плеча и избыток (+) или нехватка (-) генетического материала. Например, болезнь Дауна у мальчика: 47,XY,21 + ; синдром кошачьего крика у девочки: 46,ХХ,5р-. Молекулярно-цитогенетические методы основаны на флюоресцентной гибридизации in situ (FISH). Для исследуемой хромосомы или ее участка готовят однонитевой участок ДНК, к которому присоединяют биотин и дигоксигенин. Такой «помеченный» участок ДНК называется зондом. На микроскопическом препарате in situ денатурируют хромосомную ДНК (взятую у больного) щелочной обработкой, то есть разрывают связи между двумя цепочками ДНК. Препарат обрабатывают зондом. Так как последовательность нуклеотидов зонда и соответствующего участка исследуемой хромосомы комплементарны, то зонд присоединяется к хромосоме. В этом участке происходит ренатура.-ция ДНК. Далее препарат обрабатывают стрептовидином (вещество, избирательно присоединяющееся к биотину) и антиди-гоксигениновым антителом (избирательно присоединяется к ди-гоксигенину). К этим веществам присоединяют флюоресцентные красители (родамин — красный цвет или флюоресцеин — зеленый цвет). В люминесцентном микроскопе хорошо видны окрашенные хромосомы на фоне неокрашенных. С помощью метода FISH можно определять локализацию генов в хромосомах и все хромосомные аберрации. В последнее время разработана методика применения этого метода для диагностики анеуплоидий в интерфазных ядрах. Биохимические методы. Биохимические методы основаны на изучении активности ферментных систем либо по активности самого фермента, либо по количеству конечных продуктов реакции, катализируемой данным ферментом. Применяются хроматографические, флюорометрические, радиоиммунологические и некоторые другие методы. Они позволяют выявлять генные мутации — причины болезней обмена веществ (например, фенилкетонурии, серповидно-клеточной анемии). Они могут применяться и как экспресс-методы. С помощью биохимических нагрузочных тестов можно выявлять гетерозиготных носителей патологических генов, например фенилкетонурии. Обследуемому человеку вводят внутривенно определенное количество аминокислоты фенилаланина и через равные промежутки времени определяют его концентрацию в крови. Если человек гомозиготен по доминантному гену (АА), то концентрация фенилаланина в крови довольно быстро возвращается к контрольному уровню (определяется до введения фенилаланина), а если он гетерозиготен (Аа), то снижение концентрации фенилаланина идет в два раза медленнее. Аналогично проводятся тесты, выявляющие предрасположенность к сахарному диабету, гипертонии и другим болезням. Молекулярно-генетические методы. Эти методы позволяют анализировать фрагменты ДНК, находить и изолировать отдельные гены и их сегменты и устанавливать в них последовательность нуклеотидов. Метод клонирования ДНК позволяет изолировать отдельные гены или их части, создавать неограниченное количество их копий, транскрибировать и транслировать изолированные гены, что стало возможным благодаря открытию ферментов-рестрик-таз. Эти ферменты «узнают» специфическую олигонуклеотидную последовательность в двухнитевой ДНК и разрезают ее в данном месте — сайте. Разные рестриктазы распознают различные последовательности нуклеотидов и разрезают ДНК в разных сайтах. В последние годы для получения достаточного количества фрагментов ДНК используется полимеразная цепная реакция — метод амплификации ДНК в условиях in vitro. В течение нескольких часов можно размножить ДНК в количестве, превышающем исходное в миллионы раз. Методы гибридизации нуклеиновых кислот. После разрезания ДНК на фрагменты рестриктазами проводится их электрофорез на агарозном или полиакриламидном геле с целью разделения этих фрагментов. Далее осуществляется идентификация фрагментов ДНК. Для выявления специфических фрагментов ДНК используется метод блот-гибридизации по Саузерну. Эта методика состоит из следующих этапов: 1) после окончания электрофореза гели помещают в щелочной раствор для денатурации фрагментов ДНК — получают одноцепочечные ДНК; Этот метод позволяет обнаружить единственный ген среди десятков тысяч. Гомологичные последовательности можно идентифицировать как полностью, так и частично. Различные модификации этого метода позволяют в клинике анализировать очень малые количества ДНК, взятые у больного. Для успешного применения в практическом здравоохранении молекулярно-генетических методов необходимо создание библиотек радиоактивных зондов всех последовательностей ДНК генома человека, и в этом направлении уже немало сделано. Методы генетики соматических клеток. Наибольший интерес для генетики человека представляет метод гибридизации клеток. В 1960 г. французский ученый Ж. Барский, выращивая в культуре клетки двух линий мышей, обнаружил, что некоторые из них по своим морфологическим и биохимическим свойствам оказались промежуточными между исходными родительскими клетками. Это были гибридные клетки. Такое спонтанное слияние соматических клеток в культуре ткани происходит довольно редко. В дальнейшем было установлено, что при введении в культуру клеток РНК-содержащего вируса парагриппа Сендай, инактивированного ультрафиолетом, частота гибридизации клеток значительно повышается. В смешанной культуре разных типов клеток образуются гетерокарионы — клетки, содержащие два ядра разных клеток в одной цитоплазме. Часть таких клеток способна размножаться митозом. После митоза из двуядерного гетерокариона образуются две одноядерные клетки, каждая из которых представляет собой синкарион — настоящую гибридную клетку, содержащую хромосомы обеих родительских клеток, то есть происходит объединение двух геномов. Гибридизация возможна между клетками не только организмов разных видов (человек — мышь), но и разных типов (человек — комар). Синкарионы обычно удается получать при гибридизации в пределах класса. Например, гибридные клетки человека и мыши имеют 43 пары хромосом: 23 — от человека и 20 — от мыши. В дальнейшем происходит постепенное удаление хромосом того организма, клетки которого имеют более медленный темп размножения. У гибридных клеток человека — мыши удаляются хромосомы человека. В гибридных клетках функционируют хромосомы как человека, так и мыши, гены которых детерминируют синтез соответствующих белков. Морфологически можно отличить каждую из хромосом (дифференциальное окрашивание). Если в гибридной клетке отсутствует какая-либо хромосома и не происходит синтез каких-то белков, то можно предположить, что гены, детерминирующие синтез этих белков, локализованы в ней. Таким образом, этот метод позволяет устанавливать группы сцепления у человека, а используя нехватки и транслокации, — выяснять и последовательность расположения генов, то есть строить генетические карты хромосом человека. Экспресс-методы — это быстрые предварительные методы изучения генетики человека. Они часто используются для исследования больших контингентов людей с целью выявления наследственной патологии как скрининг-методы, применяемые при проведении просеивающих программ. Например, скрининг новорожденных на фенилкетонурию, гипотиреоз, беременных на альфа-фетопротеин, при помощи которого можно пренаталь-но определить у плода некоторые пороки развития (например, анэнцефалию, открытые формы спинномозговых грыж, синдром Дауна). К этим методам предъявляются определенные требования: 1) метод должен быть диагностически значимым, то есть положительные и отрицательные результаты должны соответствовать наличию или отсутствию заболевания; Микробиологический ингибиторный тест Гатри позволяет выявлять некоторые биохимические нарушения у новорожденных. Из пятки новорожденного берут кашпо крови на диски фильтровальной бумаги, которые помещают на агаровую культуру В. subtillis. Последнюю выращивают на минимальной питательной среде, содержащей антиметаболит искомой аминокислоты (например, фенилаланина). Антиметаболит должен одновременно тормозить рост микроба. При наличии в крови младенца большого количества фенилаланина антиметаболит разрушается и микробы начинают бурно расти. Меняя антиметаболиты, можно диагностировать наличие в крови определенных аминокислот и углеводов (лейцина, гистидина, фруктозы, галактозы и др.). Биохимические и микробиологические экспресс-методы (флюорометрические, хроматографические, радиоиммунологические и др.) широко используются для быстрой предварительной диагностики наследственных болезней обмена веществ. Выявление Х- и Y-хроматина чаще осуществляется посредством соскоба клеток слизистой оболочки щеки (буквальный эпителий). Для выявления Х-хроматина мазки окрашивают ацеторсеином (или любой другой ядерной краской) и препараты просматривают в обычном световом микроскопе. Этот метод позволяет определить количество Х-хромосом в кариотипе по количеству телец Барра (их на одну больше, чем количество глыбок Х-хроматина). Для выявления Y-хроматина мазки окрашивают 0,005 % раствором акрихин-иприта и просматривают в люминесцентный микроскоп — Y-хромосома дает яркое зеленое свечение. Этот метод позволяет установить количество Y-хромосом в кариотипе. Методы пренаталъной диагностики наследственных болезней. Пренатальная диагностика связана с решением ряда биологических и этических проблем до рождения ребенка, так как при этом речь идет не об излечении болезни, а о предупреждении рождения ребенка с патологией, не поддающейся лечению (обычно путем прерывания беременности с согласия женщины). При современном уровне развития пренатальной диагностики можно установить диагноз всех хромосомных болезней, большинства врожденных пороков развития, энзимопатий, при которых известен биохимический дефект. Часть из них можно установить практически в любом сроке беременности (хромосомные болезни), часть — после 12-й недели (редукционные пороки конечностей, атрезии, анэнцефалию), часть — только во второй половине беременности (пороки сердца, почек). Рекомендуемые методы и тесты во время беременности
Показания для пренатальной диагностики: наличие в семье точно установленного наследственного заболевания; возраст матери старше 37 лет; носительство матерью гена Х-сцепленно-го рецессивного заболевания; наличие в анамнезе у беременных спонтанных абортов в ранние сроки беременности, мертворож-дений неясного генеза, детей с множественными пороками развития и с хромосомной патологией; наличие структурных перестроек хромосом (особенно транслокаций и инверсий) у одного из родителей; гетерозиготность обоих родителей по одной паре аллелей при патологии с аутосомно-рецессивным типом наследования; беременные из зоны повышенного радиационного фона. В настоящее время применяются непрямые и прямые методы пренатальной диагностики. При непрямых методах обследуют беременную (акушерско-гинекологические методы, сыворотка крови на альфа-фетопротеин); при прямых — плод. К прямым неинвазивным (без хирургического вмешательства) методам относится ультрасонография; к прямым инвазивным (с нарушением целостности тканей) — хорионбиопсия, амниоцентез, кордоцентез и фетоскопия. Ультрасонография (эхография) — это использование ультразвука для получения изображения плода и его оболочек, состояния плаценты. Начиная с 5-й недели беременности можно получить изображение оболочек эмбриона, а с 7-й недели — и его самого. К концу 6-й недели беременности можно зарегистрировать сердечную деятельность эмбриона. В первые два месяца беременности ультразвуковое исследование еще не позволяет выявить аномалии развития плода, но можно определить его жизнеспособность. Во 2-м триместре беременности возможности ультразвуковой диагностики значительно возрастают. На 12— 20-й неделе беременности уже возможна диагностика близнецовой беременности, локализации плаценты, анэнцефалии, дефектов костной системы и закрытия невральной трубки, атрезии желудочно-кишечного тракта. По общему мнению, метод безопасен, поэтому продолжительность исследования не ограничена, и в случае необходимости его можно применять повторно. При физиологическом течении беременности целесообразно проводить двукратное УЗИ, а при беременности с высоким риском осложнений оно проводится повторно с интервалами в 2 нед. УЗИ плода обязательно при: 1) наличии у родителей и ближайших родственников врожденных пороков развития; Ультразвуковое исследование позволяет получить данные о размерах плода (длина туловища, бедра, плеча, бипариеталь-ный диаметр головы), о наличии у него дисморфии, о функции миокарда, об объеме амниотической жидкости и размерах плаценты. УЗИ позволяет обнаружить у плода ряд аномалий развития — анэнцефалию, гидроцефалию, поликистоз или агенезию почек, дисплазию конечностей, гипоплазию легких, множественные врожденные пороки, пороки сердца, водянку (отек) плода и плаценты. Допплеровское ультразвуковое сканирование (а также цветная допплерометрия) отражает кровообращение в различных тканях плода. Эхография плаценты позволяет установить ее расположение, наличие отслойки ее отдельных участков, кисты, кальцификаты (признак «старения» плаценты). Истончение или утолщение плаценты свидетельствует о вероятности фетоплацентарной недостаточности. В последние годы получает распространение ЯМР-томогра-фия плода, позволяющая выявить структурные аномалии, не обнаруживаемые при УЗИ (малые аномалии мозга, туберозный склероз, поликистоз почек и др.). Наиболее широкое распространение получила триада методов исследования: исследование уровня альфа-фетопротеина, содержания хорионического гонадотропина (ХГ) и свободного эстриола в крови женщин во 2-м триместре беременности. Содержание альфа-фетопротеина определяется также в амниоти-ческой жидкости, а свободный эстриол — в моче беременных. Отклонения плазматического уровня альфа-фетопротеина, хорионического гонадотропина, свободного эстриола у беремен-, ной служат индикаторами высокого риска для плода. Нормальные значения для определенных сроков гестации приводятся в табл. 104. Пороговыми (указывающими на высокий риск) считаются уровни альфа-фетопротеина и ХГ в крови беременной, превышающие 2МоМ (Multiple of Mediane), а для сниженного уровня альфа-фетопротеина при болезни Дауна пороговая величина менее 0,74 МоМ. Снижение уровня свободного эстриола, соответствующее значению 0,7 МоМ и ниже, также принимается как пороговое, свидетельствующее о фетоплацентарной недостаточности. Значение МоМ для белка беременности I и белка, ассоциированного с беременностью А, при наличии у эмбриона болезни Дауна или трисомии 18 составляет 0,5—0,25 МоМ. Концентрация альфа-фетопротеина определяется радиоиммунными методами. Альфа-фетопротеин обнаруживается в амниотической жидкости уже на 6-й неделе беременности (1,5 мкг/мл); наиболее высокая его концентрация наблюдается на 12—14-й неделе (около 30 мкг/мл); затем она резко снижается и на 20-й неделе составляет лишь 10 мкг/л. Хорошие результаты дает определение уровня альфа-фетопротеина в сыворотке крови матери. Его повышение обусловлено поступлением этого белка из сыворотки крови плода через плаценту при некоторых пороках развития. Состав амниотической жидкости в различные сроки беременности
При скрининг-исследованиях высокое содержание альфа-фе-топротеина в крови выявляется у 2—4 % женщин, его снижение — у 2,5 % женщин во 2-м триместре беременности. Все беременные с измененным содержанием альфа-фетопротеина в крови нуждаются в дополнительном обследовании. Содержание альфа-фетопротеина в биологических жидкостях повышено при множественных пороках развития, спинномозговой грыже, гидроцефалии, анэнцефалии, пороках развития желудочно-кишечного тракта и дефектах передней брюшной стенки, гидронефрозе и агенезии почек, а также при фетоплацентарной недостаточности, задержке внутриутробного развития плода, многоплодной беременности, преэклампсии, резус-конфликте и вирусном гепатите В. В случаях хромосомных болезней у плода (например, болезнь Дауна) или наличия у беременной сахарного диабета I типа, напротив, концентрация альфа-фетопротеина в крови беременных снижена. В настоящее время исследование альфа-фетопротеина проводится в 1-м триместре беременности одновременно с определением специфического для беременных белка А. Это позволяет диагностировать болезнь Дауна и некоторые другие хромосомные аномалии у плода уже на 11 — 13-й неделях гестации. Хорионический гонадотропин, синтезируемый трофобластом плаценты, определяется уже на 8—9-й дни после зачатия. При исследовании крови женщины во 2-м триместре беременности повышение уровня ХГ и его свободных бета-субъединиц более 2 МоМ свидетельствует о задержке внутриутробного развития плода, высоком риске антенатальной гибели плода, отслойке плаценты или о других видах фетоплацентарной недостаточности. Содержание свободного эстриола в крови беременной прогрессивно повышается за счет усиленного его синтеза плацентой из дегидроэпиандростерона, образуемого плодом, поэтому содержание свободного эстриола в биологических жидкостях (кровь, моча, амниотическая жидкость) отражает фетоплацен-тарную активность и снижается при патологии плода и нарушении функции плаценты. Исследование уровня белка беременности I (Schwangerschaft protein I) в плазме крови женщин уже в 1-м триместре беременности может служить индикатором хромосомных болезней плода, при которых он снижается. Хорионбиопсия — взятие эпителия ворсинок хориона для исследования — проводится трансцервикально (через канал шейки матки) или трансабдоминально под контролем ультра-сонографии между 8-й и 10-й неделями гестации. Полученная ткань используется для цитогенетических и биохимических исследований и анализа ДНК. С помощью этого метода можно выявлять все виды мутаций (генные, хромосомные и геномные). Значительным преимуществом биопсии ворсин хориона является то, что этот метод пренатальной диагностики может быть использован уже на ранних этапах развития плода. Если выявляются какие-либо отклонения в развитии плода и родители решают прервать беременность, то прерывание беременности на 12-й неделе (срок проведения биопсии ворсин хориона) менее опасно, чем на 18—20-й неделе, когда становятся известны результаты амниоцентеза. Амниоцентез — получение амниотической жидкости и клеток плода для последующего анализа. Это исследование стало возможным после того, как была разработана технология трансабдоминального или трансвагинального амниоцентеза, проводимого под контролем УЗИ. Получение исследуемого материала (клетки и жидкость) возможно на 16-й неделе беременности. Основные показания для амниоцентеза: общие: 1) возраст беременной более 35 лет; отдельные: 1) мертворождения, перинатальная смертность в акушерском анамнезе; Осложнения при этом методе исследования не превышают 1 %. Амниотическая жидкость используется для биохимических исследований (выявляются генные мутации), а клетки — для анализа ДНК (выявляются генные мутации), цитогенетическо-го анализа и выявления Х- и Y-хроматина (диагностируются геномные и хромосомные мутации). Простые биохимические исследования амниотической жидкости могут дать ценную диагностическую информацию — исследования содержания билирубина, эстриола, креатинина, кортизола, 17-оксипрогестерона, соотношения содержания лецитина и сфингомиелина. Диагностика адреногенитального синдрома у эмбриона (недостаточность 21-гидроксилазы) возможна уже на 8-й неделе гестации, когда в амниотической жидкости обнаруживается повышенное содержание 17-гидроксипрогестерона. Исследование спектра аминокислот амниотической жидкости позволяет выявить некоторые наследственные болезни обмена веществ у плода (аргинин-янтарную ацидурию, цитрулли-нурию и др.), а определение спектра органических кислот используется для диагностики органических ацидурий (пропионовая, метилмалоновая, изовалериановая ацидурия и др.). Для распознавания тяжести гемолитической болезни у плода при резус-сенсибилизации беременной производится прямое спект-рофотометрическое исследование амниотической жидкости. Исследование клеток амниона в культуре применяется для выявления хромосомных аберраций или определения активности ферментов. Кордоцентез — взятие крови из пуповины, клетки и сыворотка которой используются для цитогенетических, молекуляр-но-генетических и биохимических исследований. Эта процедура проводится в срок с 18-й по 22-ю неделю беременности под контролем УЗ И. Кордоцентез может быть осуществлен также при проведении эмбриофетоскопии. Процедура успешна с первой попытки в 80—97 % случаев. Преимущество кордоцентеза по сравнению с амниоцентезом заключается в том, что лимфоциты культивируются значительно быстрее и надежнее, чем амнио-циты. Например, определение вирусоспецифической ДНК или РНК (методом обратной транскрипции) в крови плода имеет решающее значение для диагностики внутриутробных инфекций — ВИЧ, краснухи, цитомегалии, парвовируса В19. Однако показания для проведения кордоцентеза ограничены в связи с высоким риском осложнений, таких как внутриутробная гибель плода (до 6 %), недонашивание беременности (9 %). Фетоскопия — осмотр плода фиброоптическим эндоскопом, введенным в амниотическую полость через переднюю стенку матки. Метод позволяет осмотреть плод, пуповину, плаценту и произвести биопсию. Фетоскопия сопровождается высоким риском прерывания беременности и технически сложна, поэтому имеет ограниченное применение. Современные технологии позволяют осуществлять биопсию кожи, мышц, печени плода для диагностики генодерматозов, мышечных дистрофий, гликогенозов и других тяжелых наследственных заболеваний. Риск прерывания беременности при применении инвазивных методов пренатальной диагностики составляет 1—2%. Везикоцентез, или пункция мочевого пузыря плода, используется для получения его мочи для исследования в случаях серьезных заболеваний и пороков развития органов мочевой системы. Доимплантационная диагностика серьезных наследственных болезней стала возможной в последнее десятилетие благодаря разработке технологии экстракорпорального оплодотворения и использования полимеразной цепной реакции для получения множественных копий эмбриональной ДНК. На стадии дробления оплодотворенной яйцеклетки (бластоцисты), когда зародыш состоит из 6—8 отдельных клеток, методами микроманипуляции отделяется одна из них для выделения ДНК, ее мультипликации и последующего анализа с помощью ДНК-зондов (прай-мерная полимеразная цепная реакция, Sauthern-blot, исследование полиморфизма рестрикционных фрагментов ДНК и др.). Эта технология применена для выявления наследственных болезней — Тея-Сакса, гемофилии, миодистрофии Дюшенна, фрагильной Х-хромосомы и ряда других. Однако она доступна немногим крупным центрам и отличается очень высокой стоимостью исследования. Разрабатываются методы выделения клеток плода (эритро-бластов, трофобластов и др.), циркулирующих в крови беременной, для проведения цитогенетических, молекулярно-генетических и иммунологических анализов в диагностических целях. Пока такая диагностика возможна лишь в тех случаях, когда в клетках крови (эритробластах) беременной имеются хромосомы или гены плода, например Y-хромосома, ген резус-фактора у резус-отрицательной женщины, антигены системы HLA, унаследованные от отца. Дальнейшее развитие и распространение методов пренаталь-ной диагностики наследственных заболеваний позволят значительно снизить частоту наследственной патологии новорожденных. ГЕННЫЕ БОЛЕЗНИ. Генные мутации у человека являются причинами многих форм наследственной патологии. Генные болезни — это разнородная по клиническим проявлениям группа заболеваний, обусловленных мутациями на генном уровне. В настоящее время описано более 3 тыс. таких наследственных болезней. Генные болезни чаще всего проявляются наследственными дефектами обмена веществ — ферментопатиями. Выделяются мутации структурных и функциональных генов. Мутации структурных генов подразделяются на: 1) сдвиг рамки считывания — вставка (инсерция) или выпадение (делеция) одной или нескольких пар нуклеотидов; в зависимости от места вставки или выпадения изменяется меньшее или большее число кодонов; Мутации функциональных генов заключаются в изменении нетранскрибируемой части молекулы ДНК, что вызывает нарушение регуляции работы структурных генов. Это может приводить к снижению или повышению скорости синтеза соответствующего белка в разной степени. Таким образом, при генных болезнях наблюдаются два вида изменения белковых продуктов. Первая группа болезней связана с качественными изменениями белковых молекул, то есть с наличием у больных аномальных белков (например, аномальные гемоглобины), обусловленными мутациями структурных генов. Другая группа заболеваний характеризуется количественными изменениями содержания нормального белка в клетке (повышенное, пониженное), что вызвано чаще всего мутациями функциональных генов. Одна и та же генная болезнь может быть обусловлена разными мутациями. Например, в гене муковисцидоза описано свыше 200 таких мутаций, в гене фенилкетонурии — 30. В некоторых случаях мутации в разных частях одного гена могут приводить к различным болезням (например, мутации RET-онкогена). Генные болезни наследуются по законам Менделя в том случае, когда речь идет о полных формах, обусловленных гамети-ческими мутациями. Это могут быть новые или унаследованные от предыдущих поколений мутации. В этих случаях патологические гены присутствуют во всех клетках организма. Однако генные мутации могут возникнуть в одной из клеток на разных стадиях дробления зиготы, и тогда организм будет мозаичен по данному гену. В одних клетках у него будет функционировать нормальная аллель, а в других — мутантная. Если эта мутация доминантная, то она проявится фенотипически в соответствующих клетках и может привести к развитию болезни (вероятно, менее тяжелой формы, чем у полных мутантов). Проблема мозаичных форм генных болезней и в генетическом, и в клиническом плане исследована недостаточно. Первичные эффекты мутантных генов могут проявляться в 4 вариантах: 1) отсутствие синтеза полипептида, Патологические мутации могут реализовываться в разные периоды онтогенеза. Большая часть их проявляется внутриутробно (до 25 % всей наследственной патологии) и в допубертатном возрасте (45 %). Около 25 % патологических мутаций проявляются в пубертатном и юношеском возрасте, и лишь 10 % моногенных болезней развиваются в возрасте старше 20 лет. Вещества, накапливающиеся в результате отсутствия или снижения активности ферментов, либо сами оказывают токсическое действие, либо включаются в цепи вторичных обменных процессов, в результате которых образуются токсические продукты. Общая частота генных болезней в популяциях людей составляет 2—4 %. Генные болезни классифицируются: согласно типам наследования (аутосомно-доминантные, аутосомно-рецессивные, Х-сцепленные доминантные и т.д.); в зависимости от системы или органа, наиболее вовлеченного в патологический процесс (нервные, глазные, кожные, эндокринные и др.); по характеру метаболического дефекта (болезни, связанные с нарушением аминокислотного, углеводного, липидного, минерального обменов, обмена нуклеиновых кислот и др.). Самостоятельную группу составляют наследственно обусловленные заболевания, возникающие при несовместимости матери и плода по антигенам групп крови. В этом случае наблюдается гемолитическая болезнь новорожденных. Типичным примером антигенной несовместимости матери и плода является несовместимость по резус-фактору. Наследование резус-фактора обусловлено тремя парами тесно сцепленных генов, расположенных в коротком плече 1-й хромосомы (1р36.2-р34), — С, D, К, имитирующих моногенное наследование. Резус-положительный фактор (Rh+) обусловлен доминантными генами (таких людей среди европейцев 85 %), а резус-отрицательный (Rh") — рецессивными (15 %). При браке женщины, имеющей резус-отрицательную группу крови, с мужчиной резус-положительной группы крови либо все дети (если мужчина гомозиготен), либо 50 % (если мужчина гетерозиготен) будут с резус-положительной группой крови. Для организма матери белки, детерминирующие резус-положительную группу крови, являются генетически чужеродными, и в ответ на парентеральное их попадание у нее вырабатываются антитела. Так как эритроциты плода чаще всего поступают в организм матери в последние недели беременности или при родах, то первая беременность обычно заканчивается благополучно. При повторной беременности сенсибилизированный материнский организм вырабатывает антитела, проникающие через плаценту в кровь плода и разрушающие резус-положительные эритроциты. У таких детей наблюдается гемолитическая болезнь новорожденных, для лечения которой производят обменное переливание крови. С каждой новой беременностью резус-положительным плодом количество антител нарастает, в результате чего гемолитическая болезнь может развиваться в ранние сроки беременностей индуцировать гибель эмбриона или плода. Отсюда понятно, что девочкам и женщинам, имеющим резус-отрицательную группу крови, ни в коем случае нельзя переливать резус-положительную кровь, так как это может повлечь за собой бесплодие. Несовместимость матери и плода возможна и по АВО-системе групп крови. НАРУШЕНИЯ АМИНОКИСЛОТНОГО ОБМЕНА. Наиболее часто встречающимися болезнями, связанными с нарушением аминокислотного обмена, являются фенилкетонурия и альбинизм. В норме аминокислота фенилаланин (ФА) с помощью фермента фенилаланингидроксилазы превращается в аминокислоту тирозин, которая в свою очередь под действием фермента тирозиназы может превращаться в пигмент меланин. При нарушении активности этих ферментов развиваются наследственные заболевания человека фенилкетонурия и альбинизм. Фенилкетонурия (ФКУ) встречается в различных популяциях людей с частотой 1:6000—1:10 000, в Беларуси — 1:6000. Она наследуется по аутосомно-рецессивному типу; больные — рецессивные гомозиготы (аа). Мутантный ген, который отвечает за синтез фермента фенилаланингидроксилазы, картирован (12q22-q24), идентифицирован и секвенирован (определена последовательность нуклеотидов). Фенилаланин принадлежит к числу незаменимых аминокислот. Только часть ФА используется для синтеза белков; основное количество этой аминокислоты окисляется до тирозина. Если фермент фенилаланингидроксилаза не активен, то ФА не превращается в тирозин, а накапливается в сыворотке крови в больших количествах в виде фенилпировиноградной кислоты (ФПВК), которая выделяется с мочой и потом, вследствие чего от больных исходит «мышиный» запах. Высокая концентрация ФПВК приводит к нарушению формирования миелиновой оболочки вокруг аксонов в ЦНС. Дети с фенилкетонурией рождаются здоровыми, но в первые же недели жизни у них развиваются клинические проявления заболевания. ФПВК является нейро-тропным ядом, в результате чего повышаются возбудимость, тонус мышц, развиваются гиперрефлексия, тремор, судорожные эпилептиформные припадки. Позже присоединяются нарушения высшей нервной деятельности, умственная отсталость, микроцефалия. У больных наблюдается слабая пигментация из-за нарушения синтеза меланина. Выделяются три формы этого заболевания. Фенилкетонурия I имеет аутосомно-рецессивный тип наследования, вызывается мутациями гена РАН, локализованного на длинном плече 12-й хромосомы (12q24.1). Фенилкетонурия //также наследуется по аутосомно-рецессивному типу, генный дефект локализуется в коротком плече 4-й хромосомы, участке 4р15.3. Частота заболевания составляет 1:100 000. Вследствие недостаточности дигидроптеридин-редуктазы нарушается восстановление активной формы тетра-гидробиоптерина, участвующего в качестве кофактора в гидроксилировании фенилаланина, тирозина и триптофана, что приводит к накоплению метаболитов, нарушению образования предшественников нейромедиаторов катехоламинового и серотонинового ряда. В патогенезе заболевания имеет значение также снижение уровня фолатов в сыворотке крови, эритроцитах, спинномозговой жидкости. Фенилкетонурия III наследуется по аутосомно-рецессивному типу и связана с недостаточностью 6-пирувоил-тетрагидроптерина синтазы, которая участвует в синтезе тетра-гидробиоптерина из дигидронеоптерина трифосфата. Частота заболевания составляет 1:30 000. Главную роль в генезе заболевания играет дефицит тетрагидробиоптерина. Диагностика заболевания осуществляется биохимическими методами: ещё до развития клинической картины в моче определяется ФПВК, в крови — высокое содержание фенилаланина. В родильных домах обязательно проводится скрининг-тест на фенилкетонурию. Альбинизм встречается в разных популяциях с разной частотой — от 1:5000 до 1:25 000. Наиболее распространенная его форма — глазо-кожный тирозиназонегативный альбинизм — наследуется по аутосомно-рецессивному типу. Основными клиническими проявлениями альбинизма в любом возрасте являются отсутствие меланина в клетках кожи (молочно-белый ее цвет), очень светлые волосы, светло-серая или светло-голубая радужная оболочка глаз, красный зрачок, повышенная чувствительность к УФ-облучению (вызывает воспалительные заболевания кожи). У больных на коже отсутствуют какие-либо пигментные пятна, снижена острота зрения. Диагностика заболевания не представляет затруднений. НАРУШЕНИЯ ОБМЕНА УГЛЕВОДОВ. Наиболее частыми наследственными дефектами, обусловленными нарушением обмена углеводов, являются' галактоземия и мукополисахаридо-зы. Галактоземия встречается с частотой примерно 1:100 000. В основе этого заболевания лежит недостаточность фермента га-лактозо-1-фосфат-уридилтрансферазы (ГФТ), переводящего га-лактозо-1-фосфат в уридиндифосфогалактозу. Генгалактозо-1-фосфат-уридилтрансферазы локализован в околоцентромерном участке 2-й хромосомы (9р). Различные мутации этого локуса клинически проявляются вариантами галактоземии. С возрастом происходит компенсаторное увеличение активности фермента уридинфосфатгалактозопирофосфорилазы, способствующего метаболизму галактозы побочным путем. Одновременно несколько активизируется и галактозо-1-фосфат-уридилтрансфераза, причем у мальчиков в пубертатном периоде в большей степени, чем у девочек. Доказано влияние тестостерона на возрастание активности обоих ферментов, на чем основан его терапевтический эффект. Описаны случаи галактоземии с не совсем обычной клинической картиной — отсутствием поражения печени. Активность ГФТ у этих больных оказалась нормальной, но была снижена активность галактокиназы (галактоземия И). Патологические изменения были связаны с высокой концентрацией галактозы в крови и тканях. Существует описание галактоземии с дефицитом галактоэпимеразы (галактоземия III). Галактоза поступает в организм с пищей (лактозой). В результате недостаточности фермента ГФТ происходит накопление галактозы и галактозо-1-фосфата в крови и разных тканях, выделение их с мочой, накопление в хрусталике галактитола (производное галактозы). Позже происходит нарушение обмена глюкозы в печени, почках, головном мозге вследствие угнетения активности фермента фосфоглкжомутазы. В крови снижается содержание глюкозы, а в моче появляются аминокислоты (метио-нин, цистеин и др.). Заболевание развивается после рождения при вскармливании младенца молоком, с которым поступает лактоза — источник неметаболизируемой галактозы. Основными симптомами заболевания являются: желтуха новорожденных, рвота и понос, приводящие к обезвоживанию организма, постепенное развитие умственной отсталости, увеличение печени и селезенки, общая дистрофия, катаракта. При лабораторном исследовании обнаруживаются галактоза и белок в моче, снижение активности галактозо-1-фосфат-уридилтрансферазы в эритроцитах. Нелеченные больные погибают в первые месяцы жизни от сопутствующих инфекций или печеночной недостаточности, у выживших развиваются катаракта и умственная отсталость. Раннее лечение диетой (исключение из пищи лактозы) обеспечивает нормальное развитие детей. Мукополисахаридозы — группа дефектов катаболизма гликоз-аминогликанов (ГАГ) с различным типом наследования (табл. 105). Так, синдромы Гурлер и Моркио наследуются по аутосом-но-рецессивному типу, синдром Хантера — по Х-сцепленному рецессивному типу. Популяционная частота их не установлена. При этом наблюдается повышенная экскреция или внутриклеточное накопление ГАГ вследствие нарушения их расщепления, обусловленного дефектами ферментов — лизосомных гидролаз. Выделяются несколько типов мукополисахаридозов. Наиболее распространен мукополисахаридоз первого типа (синдром Гурлер), обусловленный дефицитом фермента a-L-идуронидазы (локализация гена 22qll), ответственного за катаболизм кислых мукополисахаридов. Ферментативные дефекты и клинические проявления мукополисахаридозов (МПС)
Дети с синдромом Гурлер рождаются без видимых нарушений. В первые месяцы жизни черты лица становятся грубыми, западает переносица, развиваются помутнение роговицы, тугоподвижность суставов, искривление позвоночника, увеличиваются печень и селезенка. Из-за большого языка рот открыт. На 2-м году жизни выявляются короткая шея, воронкообразная грудная клетка, паховые и пупочные грыжи, увеличение головы, губ, мелкие редкие зубы, ограничение подвижности большинства суставов, прогрессирующая умственная отсталость. Позже поражается сердце, развиваются глухота и слепота. Больные погибают обычно в возрасте до 10 лет. Диагностика основана на лабораторном исследовании активности ферментов и анализе ГАГ мочи. НАРУШЕНИЯ ОБМЕНА ЛИПИДОВ. Наследственные болезни обмена липидов — обширная группа различных по генезу состояний, патогенетически связанных с нарушением жирового обмена, в том числе — плазматические липидозы, для которых характерно накопление липидных субстанций внутри клеток (болезнь Ниманна — Пика и др.), нарушения обмена ли-попротеидов (а- и р-липопротеинемии и др.). Гликолипидозы — группа заболеваний, обусловленных нарушением распада гликолипидов (цереброзидов, ганглиозидов). Гликолипидозы в основном наследуются по аутосомно-рецессивно-му типу, за исключением синдрома Фабри, ферментный дефект при котором связан с Х-хромосомой. Гликолипидозы подразделяются на цереброзидозы (болезнь Гоше, болезнь Краббе), суль-фатидозы (метахроматическая лейкодистрофия), церамидолиго-зидозы (церамидлактозидлипоидоз, синдром Фабри), ганглио-зидозы (От2-ганглиозидозы: болезнь Тея—Сакса, болезнь Занд-гоффа—Яцкевича—Пильца, болезнь Бернгеймера—Зайтельбер-га, инфантильный ганглиозидоз ГУ типа; От,-ганглиозидозы: болезнь Нормана—Ландинга, болезнь Дерри). В основе болезни Гоше лежит утрата активности фермента' глюкоцереброзидазы, приводящая к накоплению в клетках ретикулоэндотелиальной системы глюкоцереброзидов. Выделяются три типа болезни: детский (инфантильный) тип с выраженными расстройствами нервной системы (повышение тонуса мышц, приводящее в поздних стадиях к опистотонусу, клонико-тоническим судорогам, тризму) развивается в возрасте около 6 мес; хронический (тип 1), для которого характерны нарушения со стороны нервной системы; юношеский, или под-острый, тип, по тяжести течения занимает промежуточное положение. Основными симптомами заболевания являются отставание в физическом развитии, инфантилизм, увеличение печени и селезенки, последняя достигает огромных размеров, занимая почти весь живот. Почти у всех больных отмечаются проявления гиперспленизма в виде анемии, лейкопении, тромбоцитопении. Наблюдаются также склонность к кровотечениям, кожные геморрагические высыпания, обильные маточные кровотечения. Инфантильный G т2-г а н гл и о з и д о з (болезнь Гея-Сакса). В основе заболевания лежит нарушение обмена ганглиозидов, сопровождающееся повышением их уровня в сером веществе мозга в 100—300 раз. Ганглиозиды накапливаются также в печени, селезенке. При болезни Тея—Сакса определяется дефицит гексозамининидазы А — одной из форм лизосомаль-ного фермента. Тип наследования аутосомно-рецессивный, локализация гена — на 15-й хромосоме (15q22-q25.1). Частота заболевания 1 : 250 000, среди евреев-ашкенази — 1 : 4000. При рождении и в первые 3—4 мес жизни дети не отличаются от здоровых сверстников. Заболевание развивается медленно, ребенок становится менее активным, теряет приобретенные навыки, утрачивает интерес к игрушкам. Психомоторные нарушения начинают развиваться у детей с 4—6 мес. Дети становятся апатичными, перестают интересоваться окружающим, у них наблюдается мышечная гипотония. К концу 1 -го года жизни развивается слепота, обусловленная атрофией зрительных нервов; интеллект снижается до уровня идиотии. Постепенно развивается полная обездвиженность, наблюдаются судороги, не поддающиеся терапии. Смерть обычно наступает в возрасте 2—4 года. Сфингомиелиноз (болезнь Ниманна-Пика) — внутриклеточный липидоз, характеризующийся накоплением в клетках ретикулоэндотелия фосфолипида сфингомиелина из-за нарушения активности сфингомиелиназы. Тип наследования — аутосомно-рецессивный. Частота заболевания составляет 1:10 000. Болезнь генетически гетерогенна: выделяются 4 варианта — А, В, С, D. При вариантах А и В отмечается дефицит сфингомиелиназы, при вариантах С и D активность энзима нормальная. Заболевание встречается только в раннем детском возрасте и характеризуется злокачественным течением. Начало заболевания сопровождается отказом от еды, 'срыгиваниями и рвотой. Затем выявляются задержка психомоторного развития и прогрессирующая гепатоспленомегалия. В дальнейшем развиваются спастический тетрапарез, глухота и слепота. Наблюдается умеренное генерализованное увеличение лимфатических узлов. Кожа приобретает коричневый оттенок. На глазном дне обнаруживается вишнево-красное пятно. При варианте болезни А (классическая форма) имеются тяжелые поражения нервной системы, которые сочетаются с выраженной гепатоспленомегалией. Вариант В отличается более поздними сроками манифестации (2— 6 лет), отсутствием неврологической симптоматики и хроническим течением. Чрезвычайной вариабельностью начала характеризуется вариант болезни С, при котором у 40% больных отмечается пролонгированная желтуха, исчезающая к 2—4 мес жизни, типична гепатоспленомегалия. Вариант D по клинической картине сходен с вариантом С, у больных часто наблюдаются миоклонические судороги. Гиперлипопротеинемии обусловлены нарушениями обмена липидов плазмы крови вследствие дефектов ферментов или клеточных рецепторов. Липиды плазмы крови представляют собой большую группу соединений, в основном жирных кислот, три-глицеридов и холестерина. Повышенное содержание липидов в плазме крови может быть мультифакториальным или моноген-но обусловленным дефектом. Частота гетерозигот моногенно обусловленных Гиперлипопротеинемии в различных популяциях составляет от 1:100 до 1:500 (в большинстве европейских стран). Первичные гиперлипидемии — гетерогенная группа наследуемых заболеваний. Описано пять отдельных подтипов гиперлипидемии (I, II, III, IV и V). Семейная гиперхолестеринемия — наиболее распространенная форма наследуемой гиперлипидемии детского возраста, частота встречаемости 1:500. При ней выявляется повышение общего холестерина и холестерина ЛНП при нормальном содержании триглицеридов. При гетерозиготном состоянии уровень общего холестерина более 8 ммоль/л, при гомозиготном — до 18—20 ммоль/л (риск развития ишемической болезни сердца (инфаркт миокарда) соответственно в возрасте 30— 40 лет и в первом десятилетии жизни). Комбинированная семейная гиперлипиде-мия — повышение уровней холестерина и триглицеридов. Встречается чаще, чем предыдущий вариант. Во взрослом возрасте рано возникает атеросклероз коронарных и периферических сосудов. Семейная гиперлипидемия I типа характеризуется увеличением количества хиломикронов и триглицеридов, низким уровнем ос- и р-липопротеидов, снижением постгепариновой липолитической активности плазмы. Клинически заболевание характеризуется болями в животе, гепатосплено-мегалией, ксантомами. Семейная гиперлипидемия II типа обусловлена повышением в плазме крови уровней р-липопротеидов, холестерина, фосфолипидов при нормальном содержании триглицеридов. Тип IIA обусловлен дефектом гена рецепторов ЛНП, возможна также модификация аполипопротеина В-100 и апопротеина ЛОНП, у больных резко увеличено содержание ЛНП. Тип ИВ характеризуется увеличением содержания ЛНП и триглицеридов. При гомозиготном состоянии наблюдается раннее развитие атеросклероза с тяжелым поражением коронарных сосудов, при гетерозиготном — умеренное изменение липидов и развитие атеросклероза в возрасте 30—40 лет. Семейная гиперлипидемия III типа характеризуется повышенным содержанием в крови ЛНП, р-липопротеидов, пре-р-липопротеидов, холестерина, фосфолипидов и триглицеридов. Гипертриглицеридемия наблюдается при диете с высоким содержанием углеводов (нарушение толерантности к глюкозе). Клинически рано выявляются ксантомы, атероматоз, выраженная коронарная болезнь. Семейная гиперлипидемия IV типа обусловлена высоким уровнем ЛОНП, пре-р-липопротеидов и триглицеридов, триглицеридемия наблюдается при повышенном содержании углеводов в диете. Клинически выявляются нарушения толерантности к глюкозе, ишемическая болезнь. Семейная гиперлипидемия V тип а обусловлена повышенным содержанием хиломикронов, ЛОНП, пре-р-липопротеидов и триглицеридов, несколько увеличен уровень холестерина. Клинически характеризуется приступами болей в животе, гепатоспленомегалией, развитием атеросклероза и ИБС, нарушением толерантности к глюкозе. Значение гиперлипопротеинемий и их моногенных форм определяется тем, что метаболизм липидов тесно связан с патогенезом атеросклероза и ишемической болезни сердца. Генетической особенностью гиперхолестеринемии с дефектом рецепторов является то, что повышенный уровень холестерина отмечается и у гетерозигот (в 2—3 раза выше нормы) и они подвержены раннему (в 35—45 лет) развитию инфарктов миокарда. Моногенные гиперлипопротеинемий встречаются у небольшой части больных атеросклерозом. НАРУШЕНИЯ ОБМЕНА ПУРИНОВ И ПИРИМИДИНОВ. Наследственные дефекты обмена пуринов и пиримидинов рассмотрим на примере синдрома Леша-Найхана. Синдром Леша—Найхана обусловлен недостаточностью фермента гипоксантинфосфорибозилтрансферазы (ГФРТ), который катализирует присоединение свободных пуриновых оснований (гуанина и гипоксантина) к нуклеотидам. Синдром встречается редко (1:300 000 рождений); тип наследования — Х-сцепленный рецессивный. Локализация гена — Xq26-q27. При недостаточности фермента ГФРТ конечным продуктом превращения пуриновых оснований является мочевая кислота. В первые месяцы жизни отмечаются задержка развития двигательных навыков, а также экстрапирамидные хореоатетоидные движения, гиперрефлексия, клонус стоп и спастичность конечностей. Болезнь сопровождается олигофренией, склонностью ребенка к самоповреждениям. Высокое содержание мочевой кислоты и ее солей, несмотря на усиленное выделение их с мочой, приводит к формированию камней в мочевыводящих путях, отложению солей мочевой кислоты в суставах. НАРУШЕНИЯ МЕТАБОЛИЗМА МЕТАЛЛОВ. Примером нарушения метаболизма металлов может служить изменение обмена меди. Болезнь Вильсона-Коновалова (гепатоцеребральная дистрофия) обусловлена генной мутацией, в результате которой развивается дефицит фермента, тормозящего синтез белка церулоплазмина. Последний, в свою очередь, обеспечивает транспорт меди в организме. Тип наследования — аутосомно-рецес-сивный. Частота заболевания — 2:100 000 новорожденных. Соединения меди играют большую роль в обменных процессах. Ионы меди входят в состав многих ферментов митохондрий, участвующих в реакциях окисления. При недостатке церулоплаз-мина повышается концентрация меди в крови и происходит накопление ее в тканях печени и мозга (в подкорковых узлах, прежде всего в чечевицеобразных ядрах) с последующей их дегенерацией. Заболевание чаще проявляется в школьном возрасте. Первыми симптомами могут быть увеличение печени и селезенки, нарушение функций печени, ЦНС, иногда почек, снижение количества эритроцитов, тромбоцитов и лейкоцитов в крови. Поражение печени сопровождается желтухой, рвотой, диспепсией, постепенно развивается цирроз. При брюшной форме болезнь протекает по типу подострой дистрофии печени и больной умирает в состоянии печеночной комы до возникновения неврологических расстройств. Поражение ЦНС проявляется снижением интеллекта, изменением поведения (эйфория, плаксивость). Появляются дрожание рук, нарастает тонус мускулатуры, возникают выраженная ригидность, амимия: движения становятся замедленными. Ригидность осложняется контрактурами. Возникают клонические и тонические судороги. Появляются насильственные движения: гиперкинезы типа атетоза, насильственный плач, смех. Типично появление по периферии роговицы зеленовато-желтого или зеленовато-коричневого пигмента (кольцо Кайзера-Флейшера). У детей наблюдаются несколько форм заболевания. Диагностика основана на выявлении сочетанного поражения нервной системы и печени, нарушений психики, насильственного плача и смеха, а также обнаружении кольца Кайзера—Флейшера. Подтверждением диагноза служат повышение концентрации меди в моче до 1000 мкг (при норме до 100 мкг), определение концентрации церулоплазмина в сыворотке крови. НАРУШЕНИЯ СВЕРТЫВАЮЩЕЙ СИСТЕМЫ КРОВИ. Гемофилия А — тяжелое заболевание, обусловленное дефектом фактора VIII свертывания крови. Встречается с частотой 1:2500 новорожденных мальчиков. Тип наследования — Х-сцепленный рецессивный. Ген расположен в длинном плече Х-хромосомы (Xq28), структура его установлена. Заболевание распознается обычно на 2—3-м году жизни, а в тяжелых случаях — при рождении (кровотечения из пупочного канатика, внутрикожные кровоизлияния). Для него характерны множественные гематомы. Преобладают кровоизлияния в крупные суставы конечностей (коленные, локтевые, голеностопные), подкожные, внутри- и межмышечные гематомы, кровотечения при травмах и хирургических вмешательствах, наличие крови в моче. Кровоизлияния в полость суставов приводят к развитию стойкой их тугоподвижности из-за остеоартрозов (развитие соединительной ткани в суставах). Гемофилия В обусловлена снижением активности фактора IX свертывания крови. Встречается в 10 раз реже, чем гемофилия А. Тип наследования — Х-сцепленный рецессивный. Ген картирован — Xq27. Клинические проявления заболевания сходны с таковыми при гемофилии А. ГЕМОГЛОБИНОПАТИИ — заболевания, связанные с нарушением структуры молекулы гемоглобина. Нормальный гемоглобин человека (НbА) состоит из двух а-цепей и двух (3-цепей. Большую часть структурных вариантов Нb составляют одиночные замены аминокислот, в основе которых лежит замена одного азотистого основания другим с изменением кода триплета. Наиболее известной формой аномалии гемоглобина является серповидно-клеточная анемия, при которой в 6-м положении (3-цепи гемоглобина глутаминовая кислота замещена валином (HbS). Эта замена обусловливает пониженную растворимость гемоглобина, и у гомозигот эритроциты приобретают серповидную форму. Гетерозиготные носители HbS в обычных условиях клинически здоровы. У гомозигот с раннего возраста развивается характерная картина хронической гипоксии и анемии, что нередко приводит к смерти. Причина анемии — преждевременный гемолиз и распад эритроцитов, обусловленный низкой растворимостью HbS. HbS плохо связывает и переносит кислород, часто обнаруживается у населения регионов с широким распространением тропической малярии, так как гетерозиготы по HbS невосприимчивы к малярии. Талассемии обусловлены мутациями глобиновых генов, приводящими к уменьшенному содержанию глобинов или полному их отсутствию. Причиной а-талассемий служат полные делеции гемоглобиновых а-генов. Таких генов четыре, и от количества отсутствующих генов зависит тяжесть заболевания. Они расположены в 16-й хромосоме — 16р13.3. При р-талассемиях имеется дефицит синтеза р-глобина. Генетические дефекты могут быть разнообразны (делеции, точечные мутации и др.). При выраженных талассемиях наблюдается гемолитическая анемия. У гетерозиготных носителей гена р-талассемий выраженные признаки анемии обычно не обнаруживаются. ДРУГИЕ МОНОГЕННЫЕ ЗАБОЛЕВАНИЯ. Адреногеншпаль-ный синдром относится к группе наследственных нарушений биосинтеза стероидных гормонов. Известно несколько разновидностей наследственных дефицитов ферментов, обеспечивающих синтез стероидов (21-гидроксилаза, 11-гидроксилаза, 17-гидроксилаза и др.). Все варианты этой наследственной патологии наследуются по аутосомно-рецессивному типу. Ген стероид-21-гид-роксилазы картирован (6р21.3). Частота дефицита 21-гидроксилазы — 1:5000 новорожденных. Недостаточность 21-гидроксилазы определяет до 90% всех случаев врожденной гиперплазии надпочечников. Известны два классических варианта этой болезни — сольтеряющая и простая вирильная форма. Другие формы встречаются значительно реже. Сольтеряющая форма характеризуется полным дефицитом фермента и проявляется нарушением солевого обмена (дефицит минералокортикоидов). В патологический процесс вовлекается ренин-ангиотензинная система. У новорожденного отмечаются срыгивание, рвота, симптомы недостаточности периферического кровообращения, сонливость, потеря массы тела. Обезвоживание вызывает повышенную жажду. При биохимических исследованиях выявляются гиперкапиемия, гипонатриемия, ацидоз. Проста вирильная форма характеризуется прогрессирующей вирилизацией, ускоренным соматическим развитием, повышенной экскрецией гормонов коры надпочечников. У новорожденных девочек при кариотипе 46,XX отмечается различная степень маскулинизации (от умеренной гипертрофии клитора до полного срастания губно-мошоночных складок с формированием мошонки и пениса). У мальчиков заболевание проявляется на 5—7-м году жизни преждевременным половым развитием. Синдром Марфана (СМ) — аутосомно-доминантная болезнь, причиной которой является мутация гена белка фибриллина (15q21). Описано более 10 вариантов мутаций в одном гене, что обусловливает крайнюю вариабельность синдрома. Семейные случаи составляют 75% всех наблюдений. Популяционная частота-1:10 000. При СМ нарушен синтез белка фибриллина и соединительная ткань обладает повышенной растяжимостью. Симптоматика СМ многосистемная и разнообразная. Наиболее специфическими для диагностики СМ являются поражения опорно-двигательного аппарата, вывих хрусталика, пороки развития сердечно-сосудистой системы, эктазия твердой мозговой оболочки. Основные нарушения скелетно-мышечной системы: арахнодак-тилия, высокий рост, длинные конечности, деформация позвоночника (сколиоз, грудной лордоз, гиперкифоз), деформация передней стенки грудной клетки (вдавленная грудь, «куриная» грудь), ненормальная подвижность суставов, плоская стопа, высокое арковидное нёбо, недоразвитие вертлужной впадины, мышечная гипотония. Основные симптомы поражения глазной системы: вывих хрусталика, миопия, отслоение сетчатки, уплощение роговицы, увеличенная длина оси глазного яблока. В сердечно-сосудистой системе обнаруживаются: аортальная регур-гитация, аневризма восходящей части аорты, расслоение аорты, пролапс митрального клапана, кальцификация митрального отверстия, дизритмия. Для заболевания характерны паховые грыжи, спонтанный пневмоторакс, аномалии развития нервной системы. Диагностические критерии СМ должны строго соблюдаться, так как ряд других врожденных дисплазий соединительной ткани могут быть приняты за СМ. Подтверждается диагноз наличием этого заболевания у родственников 1-й степени родства. Синдром Элерса—Данлоса (СЭД) — синдром дисплазий соединительной ткани с аутосомно-доминантным типом наследования (при аутосомно-рецессивном типе наследования имеет место недостаточность лизилгидроксилазы, при сцепленном с X-хромосомой типе СЭД — недостаточность лизилоксидазы). По клинике различаются пять форм заболевания, четыре из которых имеют аутосомно-доминантное наследование. Проявляется заболевание гиперэластичностью и ранимостью кожи, ее гиперпигментацией, гипермобильностью суставов, сколиозом, трав-матизацией кожи. Час%о наблюдаются пролапс митрального клапана, генерализованный периодонтит, геморрагический синдром. Возможно нарушение интеллекта. Первая и вторая формы характеризуются развернутой симптоматикой заболевания, при третьей преобладает гипермобильность суставов, при четвертой — рецидивирующие гематомы, при пятой — гиперэластичность кожи. Нейрофиброматоз (НФ) — тяжелая многосистемная болезнь с аутосомно-доминантным типом наследования. Популяцион-ная частота — 1:3500 новорожденных. Ген картирован — 17ql 1.2 — и секвенирован. Патогенез заболевания неясен. Симптоматика НФ разнообразна, в патологический процесс вовлекаются несколько систем. Диагноз НФ можно поставить при наличии у пациента не менее двух из перечисленных признаков, но при условии, что они не являются симптомами какой-нибудь другой болезни: 1) светло-коричневые пятна на коже; пигментные пятна появляются в детстве, и их число увеличивается с возрастом; 2) наличие двух и более нейрофибром любого типа или одной плексиформной нейрофибромы; их количество с возрастом увеличивается; 3) множественные, похожие на веснушки пигментные пятна в подмышечной ямке, паховой области, на других участках тела со складками; 4) костные изменения (дисплазия крыла крыловидной кости, врожденное искривление или утончение длинных трубчатых костей, ложный сустав); 5) глиома зрительного нерва; узелки Лиша (два и более) на радужной оболочке. Течение заболевания прогрессирующее, клиническая картина разнообразна. Муковисцидоз (кистозный фиброз) обусловлен мутацией гена в 7-й хромосоме (7q31-q32), его величина — 250 000 пар нуклео-тидов. Тип наследования — аутосомно-рецессивный. В настоящее время насчитывается более 600 мутаций в гене муковисцидоза, однако лишь 6 из них (de!F508, G542X. G551D, R553X, W1282X, N1303K) в странах Европы встречаются с частотой более 1%. Одной из наиболее частых является мутация delF508. Популяционная частота — 1:2500. В основе патогенеза муковисцидоза лежит нарушение транспорта ионов С1" и Na+ через мембраны эпителиальных клеток вследствие отсутствия или изменения структуры белка — трансмембранного регулятора проводимости. В настоящее время считается, что этот белок является собственно хлорным каналом. Это приводит к избыточному выведению хлоридов, следствием чего является гиперсекреция густой слизи в клетках экзокринной части поджелудочной железы, эпителия бронхов, слизистой оболочки желудочно-кишечного тракта. Муковисцидоз представляет собой множественные поражения желез внешней секреции, проявляющиеся выделением секретов повышенной вязкости, что ведет к застойным явлениям и закупорке протоков в соответствующих органах (легких, поджелудочной железе и кишечнике) с последующими воспалительными процессами и склеротическими изменениями. В потовой жидкости повышена концентрация ионов Na+ и С1~, что является основным диагностическим лабораторным тестом. Заболевание может манифестировать в любом возрасте. У плодов и новорожденных оно проявляется мекониальной непроходимостью. Наиболее частая (75—80%) — смешанная ле-гочно-кишечная форма муковисцидоза. При этой форме легочные проявления возникают на 1—2-м году жизни и характеризуются рецидивирующими пневмониями и бронхитами с последующим развитием эмфиземы легких. Кишечные проявления связаны с нарушением активности ферментов поджелудочной железы. Гнилостные процессы в кишечнике приводят к вздутию живота, появлению обильного жирного стула с резким гнилостным запахом. В некоторых сл'учаях развивается цирроз печени. Реже встречаются преимущественно легочная (15—20%) и преимущественно кишечная (5%) формы. Диагностика основана на характерной клинической картине, определении натрия и хлоридов в потовом тесте, определении гена CFTR. Ахондроплазия (хондродистрофия) обусловлена мутацией гена рецептора фактора роста фибробластов, вызывающей отклонения в активности некоторых ферментов (5-нуклеотидазы, глю-козо-6-фосфатазы), в результате чего нарушаются рост и развитие хрящевой ткани в эпифизах трубчатых костей и в основании черепа. Тип наследования — аутосомно-доминантный. Популяционная частота — 1:100 000. Не менее 80% случаев болезни обусловлено новой мутацией гена EGFR3, локализованного на 4р. Характерными признаками заболевания являются низкий рост (120—130 см у взрослых) при сохранении нормальной длины туловища, большой череп с выступающим затылком, запавшая переносица. Конечности укорочены, в основном за счет проксимальных отделов бедренной и плечевой костей, кисти широкие и короткие. Дети отстают в моторном развитии, интеллект, как правило, не страдает. Миодистрофия Дюшенна (МД) — тяжелое заболевание, проявляющееся мышечной слабостью и повышенным содержанием в плазме крови креатинфосфокиназы (в 10—100 раз). Встречается с частотой 1:3500 новорожденных мальчиков. Тип наследования — Х-сцепленный рецессивный. Ген МД картирован в области Хр21 и детально изучен, что позволяет проводить молеку-лярно-генетическую диагностику. Характерно раннее, в возрасте 3—5 лет, начало заболевания: нарастающая слабость в мышцах бедер и таза с постепенным переходом процесса на икроножные мышцы, мышцы верхнего плечевого пояса, спины, живота и др. Появляется утиная походка. Заболевание неуклонно прогрессирует, дети оказываются прикованными к постели с 10—11-летнего возраста. Наблюдается псевдогипертрофия икроножных и ягодичных мышц за счет замещения мышечной ткани соединительной и жировой. Во многих случаях развивается сгибательная мышечная контрактура бедренных и коленных суставов и суставов верхних конечностей вследствие атрофии мышц. Рано снижаются глубокие сухожильные рефлексы. Имеется тенденция к некоторому снижению умственных способностей. Продолжительность жизни больных — 20-35 лет. Синдром фрагильной (ломкой) Х-хромосомы — заболевание, характеризующееся умеренной или глубокой умственной отсталостью и рядом физических пороков развития (большие яички, большие оттопыренные ушные раковины, выпуклый лоб и выступающие челюсти). В детстве иногда проявляется гиперреактивностью или аутизмом, в подростковом возрасте характерны общая дружелюбность, застенчивость и неагрессивность, речевые аномалии. Частота встречаемости — 1:2000 новорожденных мальчиков. Данное заболевание относится к моногенным, но закономерности наследования этой болезни необычны для Х-сцепленного признака. Этим синдромом объясняют от 20 % до 40 % всех случаев тяжелой или умеренной умственной отсталости мальчиков, которым она передавалась от матери — носителя поврежденной X-хромосомы. У этих 20—40 % больных мать-носитель получила свою поврежденную хромосому не от матери, как обычно, а от вполне здорового отца. Вторая странность этой болезни — так называемый парадокс Шермана — заключается в различной степени пенетрантности мутации синдрома ломкости Х-хромосомы в зависимости от места носителя в родословной. Третья странность — среди женщин-носителей мутантной хромосомы примерно треть оказывается в различной степени пораженной заболеванием, и дети таких пораженных женщин с большей вероятностью оказываются больными, чем дети интеллектуально нормальных женщин-носителей. Мутантный ген картирован. Он расположен в Xq28. У больных в этом участке Х-хромосомы обнаружено значительное увеличение (до 1000 и более) нестабильных тринуклеотидных повторов (CGG). В этом участке происходит снижение конденсации хроматина при культивировании лимфоцитов на специальных питательных средах, лишенных фолиевой кислоты. В настоящее время возможна пренатальная диагностика этого заболевания. Этиологической терапии пока не существует. БОЛЕЗНИ С НАСЛЕДСТВЕННЫМ ПРЕДРАСПОЛОЖЕНИЕМ отличаются от типично моногенных болезней тем, что для их проявления необходимо действие определенных факторов внешней среды. Их природа двойственная. Моногенные болезни с наследственной предрасположенностью детерминируются также одним мутантным геном, но для их проявления требуется обязательное действие конкретного фактора внешней среды, который по отношению к данной болезни может рассматриваться как специфический. Эти заболевания относительно немногочисленны, они наследуются по законам Менделя, их профилактика и лечение достаточно разработаны и эффективны. Учитывая важную роль средовых факторов в проявлении этих заболеваний, их следует рассматривать как наследственно обусловленные патологические реакции на действие внешних факторов. Это может быть извращенное реагирование на фармакологические препараты (сульфаниламиды, примахин и др.), на загрязнение атмосферы (полициклические углеводороды), на пищевые вещества и добавки (лактозу, шоколад, алкоголь), на физические (холод, ультрафиолетовые лучи) и биологические (вакцины, аллергены) факторы. Полигенные болезни с наследственным предрасположением детерминируются многими генами, каждый из которых является скорее нормальным, чем измененным. Идентификация таких генов весьма затруднена. Их патогенное действие осуществляется во взаимодействии с комплексом факторов внешней среды. Это мультифакториальные болезни. Они составляют 90% хронических неинфекционных болезней различных систем и органов человека (гипертоническая болезнь, ишемическая болезнь сердца, сахарный диабет, язвенная болезнь). Относительная роль генетических и средовых факторов различна не только для конкретной болезни, но и для каждого человека. При мультифакториальных заболеваниях всегда присутствует полигенный компонент, состоящий из взаимодействующих друг с другом мутантных генов. Индивид, унаследовавший соответствующую комбинацию этих генов, переходит «порог риска»; с этого момента факторы окружающей среды определяют, разовьется ли у данного лица и в какой степени клиническое проявление болезни. Для того чтобы аналогичный синдром проявился у другого члена данной семьи, он должен унаследовать идентичную или подобную комбинацию этих генов. Так как каждый из близких родственников больного (родители, родные братья и сестры, дети) имеет половину его генов, то все они рискуют приобрести тот же полигенный синдром. Чем отдаленнее степень родства, тем меньше вероятность наследования такой же комбинации генов. Кроме того, вероятность наследования «рискованной» комбинации генов уменьшается по мере увеличения числа генов, необходимых для проявления данного признака. Поскольку точное число генов, ответственных за мультифак-ториальные признаки, неизвестно, расчет риска наследования для родственников больного индивида основывается на эмпирических оценках, то есть на прямом подсчете соотношений больных и здоровых родственников в ранее зарегистрированных семьях. Риск повторения многофакторных условий меняется от семьи к семье, и его оценка в значительной степени зависит от количества уже пораженных членов семьи и от тяжести заболевания. Чем больше число пораженных родственников и чем тяжелее заболевание, тем выше риск для остальных родственников. В последние годы установлено, что по крайней мере треть всех генных локусов содержит полиморфные аллели, различающиеся у разных индивидов. Столь большая вариация генов создает основу для различной степени генетической предрасположенности, на которую могут налагаться действия факторов внешней среды. Наиболее очевидно ассоциируются с предрасположенностью к специфическим заболеваниям генетические локусы, образующие главный генный комплекс гистосовместимости — HLA-систему (табл. 106). В коротком плече хромосомы 6 (6р23) находится четыре тесно сцепленных, но различных локуса (А, В, С, D), связанных с HLA-комплексом. Их гены детерминируют синтез белков, расположенных на поверхности мембран клеток и позволяющих иммунной системе организма отличать собственные клетки от чужеродных. В популяциях людей каждый HLA-локус состоит из многочисленных аллелей, например ло-кус HLA-B имеет 20 аллелей. Многими исследованиями показано, что определенные аллели HLA-локусов отвечают за предрасположенность организма человека к некоторым специфическим заболеваниям. Например, если человек наследует аллель В27 в локусе HLA-B, то вероятность развития у него анкилози-рующего спондилита в 121 раз выше, чем у человека, не имеющего данного гена. Тем не менее анкилозирующий спондилит остается мультифакториальным заболеванием, поскольку для его развития необходимы помимо В27 и другие аллели. По этой причине болезнь развивается не более чем у 15 % людей, унаследовавших эту аллель. Заболевания, ассоциированные с HLA-генами
Установлена зависимость частоты некоторых заболеваний от групп крови по АВО-системе. Так, обладатели 1(0) группы крови чаще других болеют язвенной болезнью желудка и двенадцатиперстной кишки, обладатели П(А) группы крови — ишемичес-кой болезнью сердца, ревматизмом, раком желудка. Для современного этапа развития медицинской генетики характерно выявление большого числа новых наследственных болезней и синдромов. Практические врачи все чаще обнаруживают значительные различия в течении семейных форм заболеваний, не укладывающиеся в классическое описание болезни. Эти наблюдения свидетельствуют о полиморфизме и наследственной гетерогенности, свойственных наследственной патологии человека. В основе этого явления часто лежит взаимодействие генов, при котором многие признаки детерминируются несколькими генами. В других случаях один ген способен оказывать влияние на развитие нескольких признаков (явление плейотропии). ХРОМОСОМНЫЕ БОЛЕЗНИ. Хромосомными болезнями (хромосомными синдромами) называются комплексы множественных врожденных пороков развития, вызываемых числовыми (геномные мутации) или структурными (хромосомные аберрации) изменениями хромосом, видимыми в световой микроскоп. Хромосомные аберрации и изменения количества хромосом, как и генные мутации, могут возникать на разных этапах развития организма. Если они возникают в гаметах родителей, то аномалия будет наблюдаться во всех клетках развивающегося организма (полный мутант). Если аномалия возникает в процессе эмбрионального развития при дроблении зиготы, кариотип плода будет мозаичным. Мозаичные организмы могут содержать несколько (2, 3, 4 и более) клеточных клонов с различными кари-отипами. Это явление может сопровождаться мозаицизмом во всех либо в отдельных органах и системах. При незначительном количестве аномальных клеток фенотипические проявления могут не обнаруживаться. Этиологическими факторами хромосомной патологии являются все виды хромосомных мутаций (хромосомные аберрации) и некоторые геномные мутации (изменения числа хромосом). У человека встречаются только 3 типа геномных мутаций: тетра-плоидия, триплоидия и анеуплоидия. Из всех вариантов анеу-плоидий встречаются только трисомии по аутосомам, полисо-мии по половым хромосомам (три-, тетра- и пентасомии), а из моносомий — только моносомия X. У человека обнаружены все типы хромосомных мутаций: де-леции, дупликации, инверсии и транслокации. Делеция (нехватка участка) в одной из гомологичных хромосом означает частичную моносомию по этому участку, а дупликация (удвоение участка) — частичную трисомию. Если транслокация (перенос части хромосомы с одной на другую) является реципрокной (взаимной) без потери участков вовлеченных в нее хромосом, то она называется сбалансированной. Она, как и инверсия (поворот участка хромосомы на 180°), не проявляется у носителя фенотипически, так как при этом сохраняется баланс генов. Однако в процессе кросинговера у носителей сбалансированных транслокаций и инверсий могут образовываться несбалансированные гаметы, то есть гаметы с частичной дисомией, или с частичной нулисомией, или с обеими аномалиями в разных участках. В норме каждая гамета моносомна (гаплоидный набор хромосом). При потере двумя акро-центрическими хромосомами коротких плеч и соединении их центромерами может образовываться одна метацентрическая хромосома. Такие транслокации называются робертсоновскими. При концевых делециях обоих плеч хромосомы (делеции тело-меров) образуется кольцевая хромосома. У индивида, унаследовавшего такие измененные хромосомы от одного из родителей, будет частичная моносомия по одному или двум концевым участкам хромосомы. Иногда может происходить поперечный, а не продольный, как обычно, разрыв хроматид в области центромер. В этом случае образуются изохромосомы, представляющие собой зеркальное отображение двух одинаковых плеч (длинных или коротких). Наличие у индивида изохромосом проявляется фенотипически, так как имеют место одновременно и частичная моносомия (по отсутствующему плечу), и частичная трисомия (по присутствующему плечу). Хромосомные болезни у новорожденных детей встречаются с частотой примерно 2,4 случая на 1000 родившихся. Большинство хромосомных аномалий (полиплоидии, гаплоидии, трисомии по крупным хромосомам, моносомий) несовместимы с жизнью — эмбрионы и плоды элиминируются из организма матери в основном в ранние сроки беременности. Хромосомные аномалии возникают и в соматических клетках с частотой около 2 %. В норме такие клетки элиминируются иммунной системой, если они проявляют себя чужеродно. Однако в некоторых случаях (активация онкогенов) хромосомные аномалии могут быть причиной злокачественного роста. Например, транслокация между 9-й и 22-й хромосомами вызывает миелолейкоз. Патогенез хромосомных болезней еще не ясен. Специфические эффекты связаны с изменением числа структурных генов, кодирующих синтез специфических белков (увеличение при три-сомиях и уменьшение при моносомиях). Полуспецифические эффекты при хромосомных болезнях могут быть обусловлены изменением числа генов, представленных и в норме многочисленными копиями (гены тРНК, рРНК, гистоновых и рибосом-ных белков и т. п.). Неспецифические эффекты хромосомных аномалий связывают с содержанием гетерохроматина, играющего важную роль в делении клеток, их росте и других физиологических процессах. Общим для всех форм хромосомных болезней является множественность поражения. Это черепно-лицевые поражения, врожденные пороки развития систем органов, замедленные внутриутробные и постнатальные рост и развитие, отставание в психическом развитии, нарушения функций нервной, иммунной и эндокринной систем. Характеристика наиболее частых трисомий
Фенотипические проявления хромосомных мутаций зависят от следующих главных факторов: 1) особенностей вовлеченной в аномалию хромосомы (специфический набор генов); В настоящее время выяснилось, что при хромосомных мутациях наиболее специфичные для того или иного синдрома проявления обусловлены изменениями небольших участков хромосом. Так, специфические симптомы болезни Дауна обнаруживаются при трисомии небольшого сегмента длинного плеча 21-й хромосомы (21q22.1), синдрома кошачьего крика — при делеции средней части короткого плеча 5-й хромосомы (5р15), синдрома Эдвардса — при трисомии сегмента длинного плеча хромосомы Окончательный диагноз хромосомных болезней устанавливается цитогенетическими методами. ТРИСОМИИ. Наиболее часто у человека встречаются трисомии по 21-й, 13-й и 18-й паре хромосом. Синдром (болезнь) Дауна (СД) — синдром трисомии 21 — самая частая форма хромосомной патологии у человека (1:750). Цитогенетически синдром Дауна представлен простой трисоми-ей (94 % случаев), транслокационной формой (4 %) или мозаи-цизмом (2 % случаев). У мальчиков и девочек патология встречается одинаково часто. Достоверно установлено, что дети с синдромом Дауна чаще рождаются у пожилых родителей. Если возраст матери 35—46 лет, то вероятность рождения больного ребенка возрастает до 4,1 %. Возможность возникновения повторного случая заболевания в семье с трисомией хромосомы 21 составляет 1—2 % (с возрастом матери риск увеличивается). Три четверти всех случаев транслокаций при болезни Дауна обусловлены мутацией de novo. 25 % случаев транслокации носят семейный характер, при этом возвратный риск гораздо выше (до 15 %) и во многом зависит от того, кто из родителей несет симметричную транслокацию и какая из хромосом вовлечена. Масса новорожденных с синдромом Дауна в среднем составляет 3167 г. Для больных характерны округлой формы голова с уплощенным затылком, узкий лоб, широкое, плоское лицо. Типичны эпикант, запавшая спинка носа, косой (монголоидный) разрез глазных щелей, пятна Брушфильда (светлые пятна на радужке), толстые губы, утолщенный язык с глубокими бороздами, выступающий изо рта, маленькие, округлой формы, низко расположенные ушные раковины со свисающим завитком, недоразвитая верхняя челюсть, высокое нёбо, неправильный рост зубов, короткая шея. Из пороков внутренних органов наиболее типичны пороки сердца (дефекты межжелудочковой или межпредсердной перегородок, фиброэластоз и др.) и органов пищеварения (атрезия двенадцатиперстной кишки, болезнь Гиршпрунга и др.). Среди больных с синдромом Дауна с более высокой частотой, чем в популяции, встречаются случаи лейкемии и гипотиреоза. У маленьких детей резко выражена мышечная гипотония, а у детей старшего возраста часто обнаруживается катаракта. С самого раннего возраста отмечается отставание в умственном развитии. Среднее значение IQ составляет 50, но чаще встречается умеренная задержка умственного развития. Средняя продолжительность жизни при синдроме Дауна значительно ниже (36 лет), чем в популяции. Синдром Патау (СП) — синдром трисомии 13 — встречается с частотой 1:6000. Имеются два цитогенетических варианта синдрома Патау: простая трисомия и робертсоновская тр шслокация. 75 % случаев трисомии хромосомы 13 обусловлено появлением дополнительной хромосомы 13. Между частотой возникновения синдрома Патау и возрастом матери прослеживается зависимость, хотя и менее строгая, чем в случае болезни Дауна. 25 % случаев СП — следствие транслокации с вовлечением хромосом 13-й пары, в том числе в трех из четырех таких случаев мутация de novo. В четверти случаев транслокация с вовлечением хромосом 13-й пары имеет наследственный характер с возвратным риском 14 %. При СП наблюдаются тяжелые врожденные пороки. Дети с синдромом Патау рождаются с массой тела ниже нормы (2500 г). У них выявляются умеренная микроцефалия, нарушение развития различных отделов ЦНС, низкий скошенный лоб, суженные глазные щели, расстояние между которыми уменьшено, микрофтальмия и колобома, помутнение роговицы, запавшая переносица, широкое основание носа, деформированные ушные раковины, расщелина верхней губы и нёба, полидактилия, флексорное положение кистей, короткая шея. У 80 % новорожденных встречаются пороки развития сердца: дефекты межжелудочковой и межпредсердной перегородок, транспозиции сосудов и др. Наблюдаются фиброкистозные изменения поджелудочной железы, добавочные селезенки, эмбриональная пупочная грыжа. Почки увеличены, имеют повышенную дольчатость и кисты в корковом слое, выявляются пороки развития половых органов. Для СП характерна задержка умственного развития. Большинство больных с синдромом Патау (98 %) умирают в возрасте до года, оставшиеся в живых страдают глубокой идиотией. Синдром Эдвардса (СЭ)— синдром трисомии 18 — встречается с частотой примерно 1:7000. Дети с трисомией 18 чаще рождаются у пожилых матерей, взаимосвязь с возрастом матери менее выражена, чем в случаях трисомии хромосомы 21 и 13. Для женщин старше 45 лет риск родить больного ребенка составляет 0,7 %. Цитогенетически синдром Эдвардса представлен простой трисомией 18 (90 %), в 10 % случаев наблюдается мозаицизм. У девочек встречается значительно чаще, чем у мальчиков, что связано, возможно, с большей жизнестойкостью женского организма. Дети с трисомией 18 рождаются с низким весом (в среднем 2177 г), хотя сроки беременности нормальные или даже превышают норму. Фенотипические проявления синдрома Эдвардса многообразны. Наиболее часто отмечаются аномалии мозгового и лицевого черепа, мозговой череп долихоцефалической формы. Нижняя челюсть и ротовое отверстие маленькие. Глазные щели узкие и короткие. Ушные раковины деформированы и в подавляющем большинстве случаев расположены низко, несколько вытянуты в горизонтальной плоскости. Мочка, а часто и козелок отсутствуют. Наружный слуховой проход сужен, иногда отсутствует. Грудина короткая, из-за чего межреберные промежутки уменьшены и грудная клетка шире и короче нормальной. В 80 % случаев наблюдается аномальное развитие стопы: пятка резко выступает, свод провисает (стопа-качалка), большой палец утолщен и укорочен. Из дефектов внутренних органов наиболее часто отмечаются пороки сердца и крупных сосудов: дефект межжелудочковой перегородки, аплазии одной створки клапанов аорты и легочной артерии. У всех больных наблюдаются гипоплазия мозжечка и мозолистого тела, изменения структур олив, выраженная умственная отсталость, снижение мышечного тонуса, переходящее в повышение со спастикой. Продолжительность жизни детей с синдромом Эдвардса невелика: 60 % детей умирают в возрасте до 3 мес, до года доживает лишь один ребенок из.десяти; оставшиеся в живых — глубокие олигофрены. АНОМАЛИИ СОЧЕТАНИЯ ПОЛОВЫХ ХРОМОСОМ. Пол будущего ребенка определяется в момент оплодотворения в зависимости от сочетания половых хромосом (XX — женский организм, XY — мужской). При нарушении течения митоза могут образовываться необычные особи — гинандроморфы. Содержание половых хромосом в разных клетках таких особей может быть разное (мо-заицизм). У человека могут быть разные случаи мозаицизма: ХХ/ХХХ, XY/XXY, ХО/ХХХ, XO/XXY и др. Степень клинического проявления зависит от количества мозаичных клеток — чем их больше, тем сильнее проявление. При нормальном течении мейоза у женского организма образуется один тип гамет, содержащих Х-хромосому. Однако при нерасхождении половых хромосом могут образовываться еще два типа гамет — XX и 0 (не содержащая половых хромосом). У мужского организма в норме образуется два типа гамет, содержащих Х- и Y-хромосомы. При нерасхождении половых хромосом возможны варианты гамет XY и 0. Рассмотрим возможные комбинации половых хромосом в зиготе у человека (их 12) и проанализируем каждый вариант. XX— нормальный женский организм. XXX— синдром трисомии X. Частота встречаемости 1:1000. Кариотип 47,ХХХ. В настоящее время имеются описания тетра-и пентосомий X. Трисомия по Х-хромосоме возникает в результате нерасхождения половых хромосом в мейозе или при первом делении зиготы. Синдрому полисемии X присущ значительный полиморфизм. Женский организм с мужеподобным телосложением. Могут быть недоразвиты первичные и вторичные половые признаки. В 75 % случаев у больных наблюдается умеренная степень умственной отсталости. У некоторых из них нарушена функция яичников (вторичная аменорея, дисменорея, ранняя менопауза). Иногда такие женщины могут иметь детей. Повышен риск заболевания шизофренией. С увеличением числа дополнительных Х-хромосом нарастает степень отклонения от нормы. ХО— синдром Шерешевского-Тернера (моносомия X). Частота встречаемости 1:2000—1:3000. Кариотип45,Х. У 55 % девочек с этим синдромом обнаруживается кариотип 45,X, у 25 % — изменение структуры одной из Х-хромосом. В 15 % случаев выявляется мо-заичность в виде двух или более клеточных линий, одна из которых имеет кариотип 45,X, а другая представлена кариотипами 46,XX или 46,XY. Третья клеточная линия наиболее часто представлена кариотипом 45,Х, 46^ХХ, 47,ХХХ. Риск наследования синдрома составляет 1 случай на 5000 новорожденных. Фенотип женский. У новорожденных и детей грудного возраста отмечаются признаки дисплазии: короткая шея с избытком кожи и крыловидными складками, лимфатический отек стоп, голеней, кистей рук и предплечий, вальгусная деформация стоп, множественные пигментные пятна, низкорослость. В подростковом возрасте выявляются отставание в росте (рост взрослых 135—145 см) и в развитии вторичных половых признаков. Для взрослых характерно низкое расположение ушных раковин, недоразвитие первичных и вторичных половых признаков, дисгенезия гонад, сопровождающаяся первичной аменореей. У 20 % больных имеются пороки сердца (коарктация аорты, стеноз аорты, пороки развития митрального клапана), у 40 % — пороки почек (удвоение мочевыводящих путей, подковообразная почка). У больных, имеющих клеточную линию с Y-хромосомой, может развиться го-надобластома, часто наблюдается аутоиммунный тиреоидит. Интеллект страдает редко. Недоразвитие яичников приводит к бесплодию. Для подтверждения диагноза наряду с исследованием клеток периферической крови проводятся биопсия кожи и исследование фибробластов. В некоторых случаях генетическое исследование позволяет выявить синдром Нуннан, который имеет схожие фенотипические проявления, однако этиологически не связан с синдромом Шерешевского—Тернера. В отличие от последнего при синдроме Нуннан заболеванию подвержены как мальчики, так и девочки, а в клинической картине доминирует задержка умственного развития, характерен Тернер-фенотип при нормальном мужском или женском кариотипе. У большинства больных синдромом Нуннан имеется нормальное половое развитие и сохранена фертильность. В большинстве случаев заболевание не сказывается на продолжительности жизни пациентов. XY— нормальный мужской организм. XXY и XXXY— синдром Клайнфелтера. Частота встречаемости 1:500. Кариотип 47,XXY у 80 % мальчиков с синдромом Клайнфелтера, в 20 % случаев обнаруживается мозаицизм, при котором одна из клеточных линий имеет кариотип 47,XXY. Возвратный риск для синдрома Клайнфелтера не превышает общепопу-ляционные показатели и составляет 1 случай на 2000 живорожденных детей. Фенотип мужской. Клиника отличается широким разнообразием и неспецифичностью проявлений. У мальчиков с этим синдромом рост превышает средние показатели, характерные для данной семьи, у них длинные конечности, женский тип телосложения, гинекомастия. Слабо развит волосяной покров, снижен интеллект. Вследствие недоразвития семенников слабо выражены первичные и вторичные половые признаки, нарушено течение сперматогенеза. Половые рефлексы сохранены. Иногда эффективно раннее лечение мужскими половыми гормонами. Чем больше в наборе Х-хромосом, тем значительнее снижен интеллект. Инфантильность и поведенческие проблемы при синдроме Клайнфелтера создают трудности социальной адаптации. YO и 00— зиготы нежизнеспособны. Иногда возможны случаи увеличения количества Y-хромо-сом: XYY, XXYY и др. При этом больные имеют признаки синдрома Клайнфелтера, высокий рост (в среднем 186 см) и агрессивное поведение. Могут быть аномалии зубов и костной системы. Половые железы развиты нормально. Чем больше в наборе Y-хромосом, тем значительнее снижение интеллекта. СИНДРОМЫ ЧАСТИЧНЫХ АНЕУПЛОИДИЙ. Помимо полных трисомий и моносомий известны синдромы, связанные с частичными трисомиями и моносомиями практически по любой хромосоме. Однако эти синдромы встречаются реже одного случая на 100 000 рождений. Синдром трисомий по короткому плечу 9-й хромосомы (9р+) — наиболее частая форма частичных трисомий (описано свыше 200 случаев). Для больных с трисомией 9р+ характерны умственная отсталость, задержка роста, микроцефалия, антимонголоидный разрез глазных щелей, глубоко посаженные глаза, опущенные уголки рта, нос с характерным округлым кончиком, низко расположенные оттопыренные ушные раковины, недоразвитие ногтей и дистальных фаланг пальцев рук. Часто наблюдаются выступающие лобные кости, повышенная обволошенность, пятна цвета кофе с молоком на коже, эпикант, косоглазие, высокое дуто-образное нёбо, короткая шея, сколиоз, частичная синдактилия пальцев стоп. Примерно в четверти случаев обнаруживаются врожденные пороки сердца. Прогноз для жизни сравнительно благоприятный — описаны больные, достигшие преклонного возраста. Синдромы частичных моносомий распространены примерно с такой же частотой, как и синдромы частичных трисомий. Наиболее известные из них — синдромы Вольфа—Хиршхорна, кошачьего крика, Орбели. Синдром Вольфа—Хиршхорна (4р-) обусловлен делецией короткого плеча хромосомы 4. Популяционная частота заболевания — около 1 случая на 100 000. Дети с синдромом Вольфа— Хиршхорна обычно рождаются у молодых родителей, доношенные, но со значительно сниженным весом (около 2000 г). Для таких детей характерна резкая задержка физического и психомоторного развития. У них наблюдаются умеренно выраженная микроцефалия, клювовидный нос, выступающее надпереносье, деформированные, низко расположенные ушные раковины, вертикальные складки кожи впереди ушных раковин, гипотония мышц, значительное снижение реакции на внешние раздражения, судорожные припадки. Отмечаются также расщелины верхней губы и нёба, деформации стоп, аномалии глазных яблок, эпикант и маленький рот с опущенными уголками. Из внутренних органов чаще поражаются сердце (пороки развития) и примерно в половине случаев — почки (гипоплазия и кисты). Большинство детей с синдромом 4р- умирает на 1-м году жизни. Максимальный известный возраст пациента с этим синдромом — 25 лет. Синдром кошачьего крика (5р-) обусловлен делецией короткого плеча 5-й хромосомы. Популяционная частота синдрома -примерно 1:45 000. Для данного синдрома наиболее характерны специфический плач, напоминающий кошачье мяуканье, лунообразное лицо, мышечная гипотония, умственное и физическое недоразвитие, микроцефалия, низко расположенные, иногда деформированные ушные раковины, эпикант, антимонголоидный разрез глазных щелей, косоглазие. Иногда наблюдаются атрофия зрительного нерва и очаги депигментации сетчатки. Как правило, выявляются пороки сердца. Наиболее постоянный признак синдрома — «кошачий крик» — обусловлен изменениями гортани: сужением, мягкостью хрящей, отечностью или необычной складчатостью слизистой оболочки, уменьшением надгортанника. Изменения других органов и систем неспецифичны. Продолжительность жизни у больных с этим синдромом значительно снижена, только около 14 % из них переживают возраст 10 лет. Синдром Орбели (13q-) обусловлен делецией длинного плеча 13-й хромосомы, сегментов 13q22-q31. Популяционная частота синдрома не установлена. Дети с синдромом Орбели рождаются с низким весом (2200 г). Клинически синдром проявляется аномалиями развития всех систем организма. Характерны микроцефалия, отсутствие носо- вой вырезки (лоб непосредственно переходит в нос), эпикант, антимонголоидный разрез глаз, широкая спинка носа, высокое нёбо, низко расположенные деформированные ушные раковины. Отмечаются поражения глаз (микрофтальмия, иногда аноф-тальмия, косоглазие, катаракта, ретинобластома), опорно-двигательного аппарата (короткая шея, гипо- или аплазия первого пальца кисти и пяточной кости, синдактилии кистей и стоп), атрезии прямой кишки и заднепроходного отверстия. Часты пороки развития сердца, почек, головного мозга. Для всех детей с синдромом Орбели характерна глубокая олигофрения, возможны потеря сознания, судороги. Большинство больных с синдромом 13q- погибают на 1-м году жизни. Синдром Прадера—Вилли— делеция хромосомы 15, у 70 % больных наблюдается делеция 15-й хромосомы (отцовская аллель), у 5 % заболевание связано с перестройкой хромосомы 15. В некоторых случаях хромосомная аномалия не идентифицируется, но у таких пациентов обнаруживается характерная клиническая картина. Основными клиническими признаками являются отставание умственного развития, поведенческие нарушения, задержка физического развития, низкорослость, гипотония. Наряду с диспластическими признаками (череп со сдавленной с боков лобной частью, миндалевидный разрез глаз, опущенные углы рта, маленькие стопы и кисти) выявляется гипогонадизм, обусловленный низким уровнем половых гормонов. Медико-генетическое консультирование — отрасль профилактической медицины, главной целью которой является снижение количества генетически обусловленных болезней и врожденных пороков развития. Появление генетической консультации как самостоятельного учреждения обычно связывают с именем S.C. Reed (1947), однако еще в 30-х годах прошлого века русский клиницист-невропатолог С.Н. Давиденков проводил генетическое консультирование и сформулировал основные положения по методике консультирования семей с наследственными заболеваниями нервной системы (1934). Современная генетическая консультация призвана служить интересам семьи и общества. Цель генетической консультации — установление степени генетического риска в обследуемой семье и разъяснение супругам в доступной форме медико-генетического заключения. Задачи медико-генетического консультирования: 1) ретро- и проспективное консультирование семей и больных с наследственной или врожденной патологией; Иначе говоря, задачей генетической консультации является составление генетического прогноза в семье индивидуума с аномалией физического, психического либо полового развития и выбор профилактических мероприятий по предупреждению рождения больного ребенка. Составление генетического прогноза включает три этапа. 1. Определение степени генетического риска. Под генетическим риском понимается вероятность (от 0 до 100 %) возникновения определенной аномалии у самого пациента (пробанда) или его родственников. Общий риск проявления генетически обусловленной аномалии для популяций европейцев составляет 3—5 % (генетический груз), поэтому риск, который не превышает 5 %, расценивается как низкий. Генетический риск до 10 % называется повышенным в легкой степени, до 20 % — повышенным в средней степени и свыше 20 % — высоким. С генетической точки зрения можно пренебречь риском, не выходящим за пределы повышенного в легкой степени, и не считать его противопоказанием к деторождению даже тогда, когда нет возможности пренатальной диагностики предполагаемой аномалии. В любом случае семья должна знать о степени генетического риска для решения вопроса о планировании беременности или ее прерывании. Врач-генетик лишь оказывает помощь в принятии такого решения. 2. Оценка тяжести медицинских и социальных последствий предполагаемой аномалии. Степень генетического риска далеко не всегда соответствует степени тяжести ожидаемой патологии. Например, полидактилия (аутосомно-доминантный тип наследования, высокая степень генетического риска — не менее 50 %) может быть легко устранена соответствующей корригирующей операцией, и человек может вести нормальный образ жизни, в то время как фенилкетонурия, риск возникновения которой у детей гетерозиготных родителей составляет 25 %, — это тяжелое заболевание, плохо поддающееся лечению. Во втором случае степень страдания с медицинской точки зрения и социальных последствий для больного и его семьи расценивается как тяжелая. 3. На третьем этапе медико-генетического консультирования врач-генетик должен оценить перспективы применения и эффективность методов пренатальной диагностики. Достижения в этой области позволяют планировать деторождение в семьях с высоким риском наследования тяжелой патологии (болезнь Дауна, мукополисахаридоз, гемофилия, муковисцидоз и др.), так как эти заболевания могут быть выявлены методами пренаталь-ной диагностики. Показания для направления семьи в медико-генетическую консультацию: - наличие сходных заболеваний у нескольких членов семьи; При медико-генетическом консультировании существует ряд трудностей морально-этического характера: вмешательство в семейную тайну (возникает при сборе данных для построения родословных, при выявлении носителей патологического гена, при несовпадении паспортного и биологического отцовства и др.; проблема разрешается корректным отношением врача к пациенту); ответственность врача-генетика в случае совета консультирующимся на основании вероятностного прогноза (необходимо, чтобы пациент правильно понял медико-генетическую информацию, консультант не должен давать категорических советов; окончательное решение принимают сами консультирующиеся). Поделитесь этой записью или добавьте в закладки | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||